药典目录
0861 残留溶剂测定法
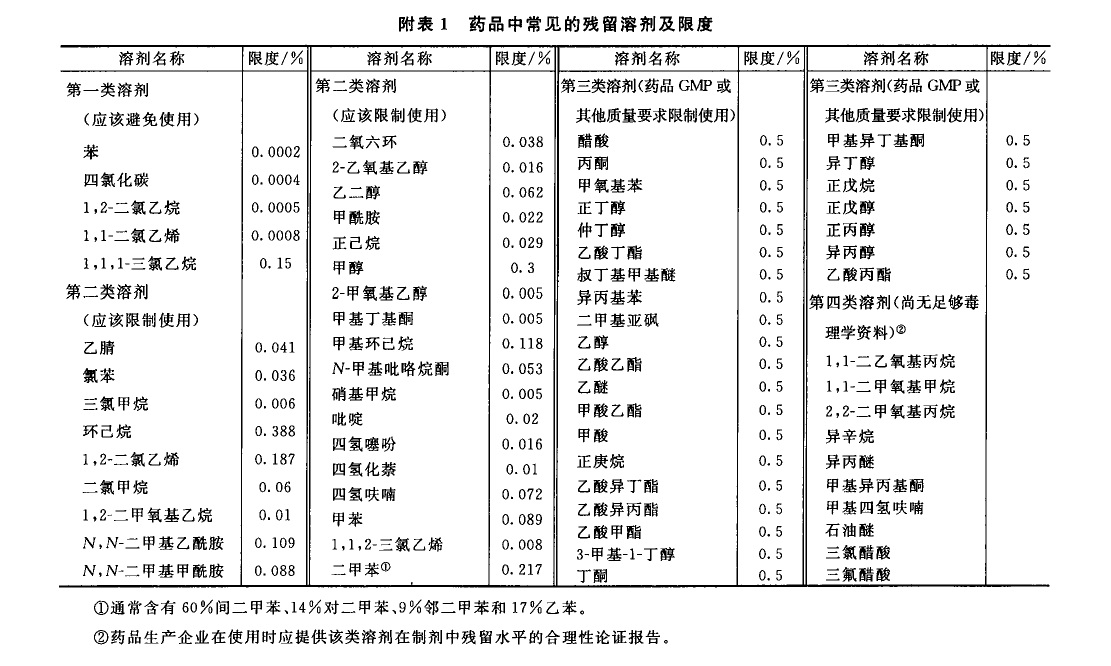
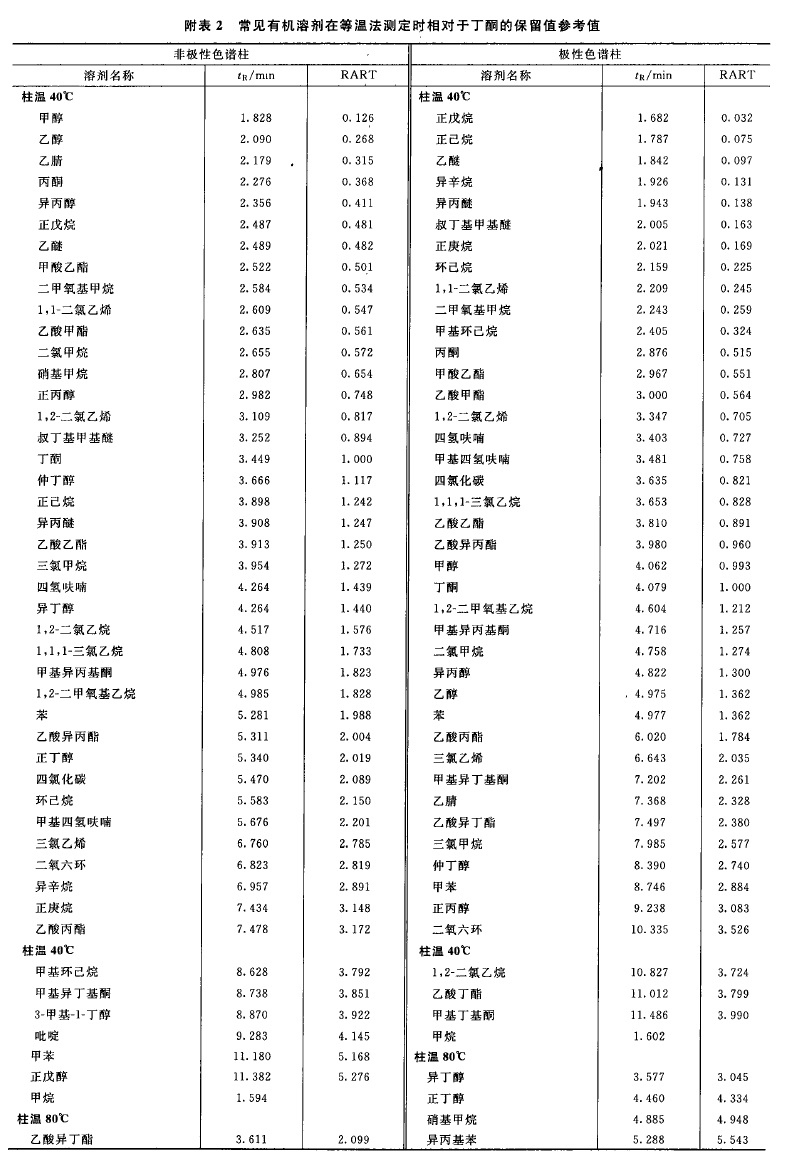
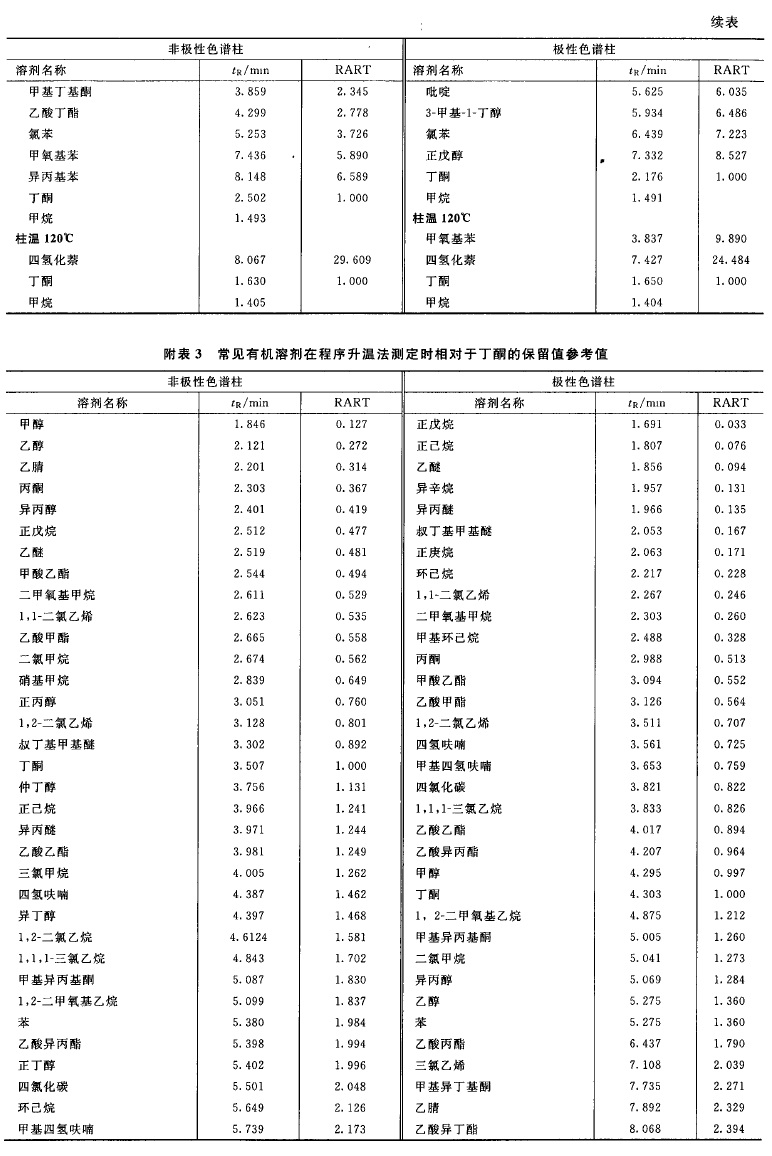
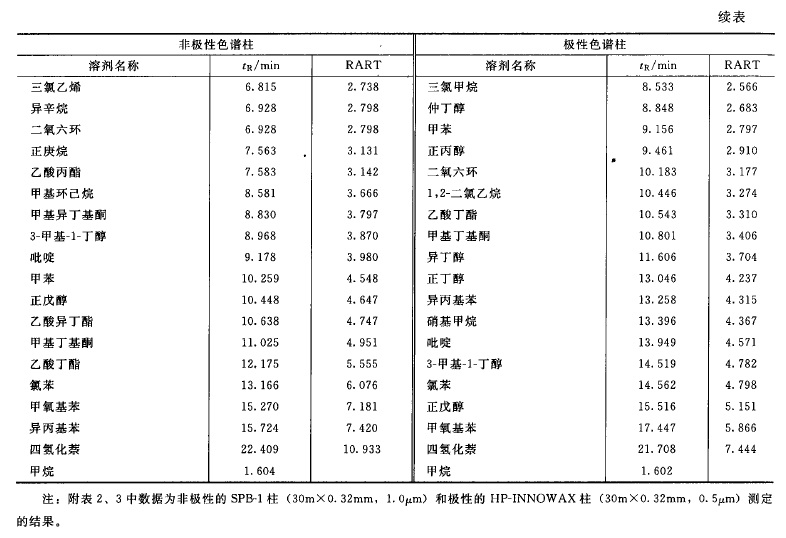
来源:三部 分类:三部通则 页码:4630861 残留溶剂测定法 药品中的残留洛剂系指在原料药或辅料的生产中,以及在制剂制备过程中使用的,但在工艺过程中未能完全去除的有机溶剂。药品中常见的残留溶剂及限度见附表1,除另有规定外,第一、第二、第三类溶剂的残留限度应符合附表1中的规定;对其他溶剂,应根据生产工艺的特点,制定相应的限度,使其符合产品规范、药品生产质量管理规范(GMP)或其他基本的质量要求。 本法照气相色谱法(通则0521)测定。 色谱柱 1.毛细管柱 除另有规定外,极性相近的同类色谱柱之间可以互换使用。 (1)非极性色谱柱 固定液为100%的二甲基聚硅氧烷的毛细管柱。 (2)极性色谱柱 固定液为聚乙二醇(PEG-20M)的毛细管柱。 (3)中极性色谱柱 固定液为(35%)二苯基-(65%)甲基聚硅氧烷、(50%)二苯基-(50%)二甲基聚硅氧烷、(35%)二苯基-(65%二甲基聚硅氧烷、(14%)氰丙基苯基-(86%)二甲基聚硅氧烷、(6%)氰丙基苯基-(94%)二甲基聚硅氧烷的毛细管柱等。 (4)弱极性色谱柱 固定液为(5%)苯基-(95%)甲基聚硅氧烷、(5%)二苯基-(95%)二甲基硅氧烷共聚物的毛细管柱等。 2.填充柱 以直径为0.18~0.25mm的二乙烯苯-乙基乙烯苯型高分子多孔小球或其他适宜的填料作为固定相。 系统适用性试验 (1)用待测物的色谱峰计算,毛细管色谱柱的理论板数一般不低于5000;填充柱的理论板数一般不低于1000。 (2)色谱图中,待测物色谱峰与其相邻色谱峰的分离度应大于1.5。 (3)以内标法测定时,对照品溶液连续进样5次,所得待测物与内标物峰面积之比的相对标准偏差(RSD)应不大于5%;若以外标法测定,所得待测物峰面积的RSD应不大于10%。 供试品溶液的制备 1.顶空进样 除另有规定外,精密称取供试品0.1~1g;通常以水为溶剂;对于非水溶性药物,可采用N,N-二甲基甲酰胺、二甲基亚砜或其他适宜溶剂;根据供试品和待测溶剂的溶解度,选择适宜的溶剂且应不干扰待测溶剂的测定。根据各品种项下残留溶剂的限度规定配制供试品溶液,其浓度应满足系统定量测定的需要。 2.溶液直接进样 精密称取供试品适量,用水或合适的有机溶剂使溶解;根据各品种项下残留溶剂的限度规定配制供试品溶液,其浓度应满足系统定量测定的需要。 对照品溶液的制备 精密称取各品种项下规定检查的有机溶剂适量,采用与制备供试品溶液相同的方法和溶剂制备对照品溶液;如用水作溶剂,应先将待测有机溶剂溶解在50%二甲基亚砜或N,N-二甲基甲酰胺溶液中,再用水逐步稀释。若为限度检查,根据残留溶剂的限度规定确定对照品溶液的浓度;若为定量测定,为保证定量结果的准确性,应根据供试品中残留溶剂的实际残留量确定对照品溶液的浓度;通常对照品溶液色谱峰面积不宜超过供试品溶液中对应的残留溶剂色谱峰面积的2倍。必要时,应重新调整供试品溶液或对照品溶液的浓度。 测定法 第一法(毛细管柱顶空进样等温法) 当需要检查有机溶剂的数量不多,且极性差异较小时,可采用此法。 色谱条件 柱温一般为40~100℃;常以氮气为载气,流速为每分钟1.0~2.0ml;以水为溶剂时顶空瓶平衡温度为70~85℃,顶空瓶平衡时间为30~60分钟;进样口温度为200℃;如采用火焰离子化检测器(FID),温度为250℃。 测定法 取对照品溶液和供试品溶液,分别连续进样不少于2次,测定待测峰的峰面积。 对色谱图中未知有机溶剂的鉴别,可参考附表2进行初筛。 第二法(毛细管柱顶空进样系统程序升温法) 当需要检查的有机溶剂数量较多,且极性差异较大时,可采用此法。 色谱条件 柱温一般先在40℃维持8分钟,再以每分钟8℃的升温速率升至120℃,维持10分钟;以氮气为载气,流速为每分钟2.0ml;以水为溶剂时顶空瓶平衡温度为70~85℃,顶空瓶平衡时间为30~60分钟;进样口温度为200℃;如采用FID检测器,进样口温度为250℃。 具体到某个品种的残留溶剂检查时,可根据该品种项下残留溶剂的组成调整升温程序。 测定法 取对照品溶液和供试品溶液,分别连续进样不少于2 次,测定待测峰的峰面积。 对色谱图中未知有机溶剂的鉴别,可参考附表3进行初筛。 第三法(溶液直接进样法) 可采用填充柱,亦可采用适宜极性的毛细管柱。 测定法 取对照品溶液和供试品溶液,分别连续进样2~3次,测定待测峰的峰面积。 计算法 (1)限度检查除另有规定外,按各品种项下规定的供试品溶液浓度测定。以内标法测定时,供试品溶液所得被测溶剂峰面积与内标峰面积之比不得大于对照品溶液的相应比值。以外标法测定时,供试品溶液所得被测溶剂峰面积不得大于对照品溶液的相应峰面积。 (2)定量测定按内标法或外标法计算各残留溶剂的量。 【附注】 (1)除另有规定外,顶空条件的选择: ①应根据供试品中残留溶剂的沸点选择顶空平衡温度。对沸点较高的残留溶剂,通常选择较高的平衡温度;但此时应兼顾供试品的热分解特性,尽量避免供试品产生的挥发性热分解产物对测定的干扰。 ②顶空平衡时间一般为30~45分钟,以保证供试品溶液的气-液两相有足够的时间达到平衡。顶空平衡时间通常不宜过长,如超过60分钟,可能引起顶空瓶的气密性变差,导致定量准确性的降低。 ③对照品溶液与供试品溶液必须使用相同的顶空条件。 (2)定量方法的验证当采用顶空进样时,供试品与对照品处于不完全相同的基质中,故应考虑气液平衡过程中的基质效应(供试品溶液与对照品溶液组成差异对顶空气-液平衡的影响)。由于标准加入法可以消除供试品溶液基质与对照品溶液基质不同所致的基质效应的影响,故通常采用标准加入法验证定量方法的准确性;当标准加入法与其他定量方法的结果不一致时,应以标准加入法的结果为准。 (3) 干扰峰的排除 供试品中的未知杂质或其挥发性热降解物易对残留溶剂的测定产生干扰。干扰作用包括在测定的色谱系统中未知杂质或其挥发性热降解物与待测物的保留值相同(共出峰);或热降解产物与待测物的结构相同(如甲氧基热裂解产生甲醇)。当测定的残留溶剂超出限度,但未能确定供试品中是否有未知杂质或其挥发性热降解物对测定有干扰作用时,应通过试验排除干扰作用的存在。对第一类干扰作用,通常采用在另一种极性不同的色谱柱系统中对相同供试品再进行测定,比较不同色谱系统中测定结果的方法。如两者结果一致,则可以排除测定中有共出峰的干扰;如两者结果不一致,则表明测定中有共出峰的干扰。对第二类干扰作用,通常要通过测定已知不含该溶剂的对照样品来加以判断。 (4)含氮碱性化合物的测定 普通气相色谱仪中的不锈钢管路、进样器的衬管等对有机胺等含氮碱性化合物具有较强的吸附作用,致使其检出灵敏度降低,应采用惰性的硅钢材料或镍钢材料管路;采用溶液直接进样法测定时,供试品溶液应不呈酸性,以免待测物与酸反应后不易汽化。 通常采用弱极性的色谱柱或其填料预先经碱处理过的色谱柱分析含氮碱性化合物,如果采用胺分析专用柱进行分析,效果更好。 对不宜采用气相色谱法测定的含氮碱性化合物,如N-甲基吡咯烷酮等,可采用其他方法如离子色谱法等测定。 (5)检测器的选择 对含卤素元素的残留溶剂如三氯甲烷等,采用电子捕获检测器(ECD ) ,易得到高的灵敏度。 (6)由于不同的实验室在测定同一供试品时可能采用了不同的实验方法,当测定结果处于合格与不合格边缘时,以采用内标法或标准加入法为准。 (7)顶空平衡温度一般应低于溶解供试品所用溶剂的沸点10℃以下,能满足检测灵敏度即可;对于沸点过高的溶剂,如甲酰胺、2-甲氧基乙醇、2-乙氧基乙醇、乙二醇、N-甲基吡咯烷酮等,用顶空进样测定的灵敏度不如直接进样,一般不宜用顶空进样方式测定。 (8)利用保留值定性是气相色谱中最常用的定性方法。色谱系统中载气的流速、载气的温度和柱温等的变化都会使保留值改变,从而影响定性结果。校正相对保留时间(RART)只受柱温和固定相性质的影响,以此作为定性分析参数较可靠。应用中通常选用甲烷测定色谱系统的死体积(t0): tR-t0 RART =---------- tˊR-t0 式中 tR为组分的保留时间; tˊR 为参比物的保留时间。