2341 农药残留量测定法
本方法系用气相色谱-串联质谱法和液相色谱-串联质谱法测定药材、饮片及制剂中部分农药残留量。除另有规定外,按下列方法测定。
第一法 有机氯类农药残留量测定法(色谱法)
1 9种有机氯类农药残留量测定法
色谱条件与系统适用性试验 以(14%-氰丙基-苯基)甲基聚硅氧烷或(5%苯基)甲基聚硅氧烷为固定液的弹性石英毛细管柱(0.32mm×30m, 0.25μm),63Ni-ECD电子捕获检测器。进样口温度230℃,检测器温度300℃,不分流进样。程序升温:初始100℃,每分钟10℃升至220℃,每分钟8℃升至250℃,保持10分钟。理论板数按α-BHC峰计算应不低于1×106,两个相邻色谱峰的分离度应大于1.5。
对照品贮备溶液的制备 精密称取六六六(BHC)(α-BHC,β-BHC,γ-BHC,δ-BHC)、滴滴涕(DDT)(p,p'-DDE,p,p'-DDD,o,p'-DDT,p,p'-DDT)及五氯硝基苯(PCNB)农药对照品适量,用石油醚(60~90℃)分别制成每1ml含4~5μg的溶液,即得。
混合对照品贮备溶液的制备 精密量取上述各对照品贮备液0.5ml,置10ml量瓶中,用石油醚(60~90℃)稀释至刻度,摇匀,即得。
混合对照品溶液的制备 精密量取上述混合对照品贮备液,用石油醚(60~90℃)制成每1L分别含0、1μg、5μg、10μg、50μg、100μg、250μg的溶液,即得。
供试品溶液的制备 药材或饮片 取供试品,粉碎成粉末(过三号筛),取约2g,精密称定,置100ml具塞锥形瓶中,加水20ml浸泡过夜,精密加丙酮40ml,称定重量,超声处理30分钟,放冷,再称定重量,用丙酮补足减失的重量,再加氯化钠约6g,精密加二氯甲烷30ml,称定重量,超声15分钟,再称定重量,用二氯甲烷补足减失的重量,静置(使分层),将有机相迅速移入装有适量无水硫酸钠的100ml具塞锥形瓶中,放置4小时。精密量取35ml,于40℃水浴上减压浓缩至近干,加少量石油醚(60~90℃)重复操作至二氯甲烷及丙酮除净,用石油醚(60~90℃)溶解并转移至10ml具塞刻度离心管中,加石油醚(60~90℃)精密稀释至5ml,小心加入硫酸1ml,振摇1分钟,离心(3000r/min)10分钟,精密量取上清液2ml,置具刻度的浓缩瓶(图1)中,连接旋转蒸发器,40℃下(或用氮气)将溶液浓缩至适量,精密稀释至1ml,即得。
图1 刻度浓缩瓶
制剂 取供试品,研成细粉(蜜丸切碎,液体直接量取),精密称取适量(相当于药材2g),按上述供试品溶液制备法制备,即得供试品溶液。
测定法 分别精密吸取供试品溶液和与之相对应浓度的混合对照品溶液各1μl,注入气相色谱仪,按外标法计算供试品中9种有机氯农药残留量。
2 22种有机氯类农药残留量测定法
色谱条件及系统适用性试验 分析柱:以50%苯基-50%二甲基聚硅氧烷为固定液的弹性石英毛细管柱(0.25mm×30m, 0.25μm),验证柱:以100%二甲基聚硅氧烷为固定液的弹性石英毛细管柱(0.25mm×30m, 0.25μm),63Ni-ECD电子捕获检测器。进样口温度240℃,检测器温度300℃,不分流进样,流速为恒压模式(初始流速为1.3ml/min)。程序升温:初始70℃,保持1分钟,每分钟10℃升至180℃,保持5分钟,再以每分钟5℃升至220℃,最后以每分钟100℃升至280℃,保持8分钟。理论板数按α-BHC计算应不低于1×106,两个相邻色谱峰的分离度应大于1.5。
对照品贮备溶液的制备 精密称取表1中农药对照品适量,用异辛烷分别制成如表1中浓度,即得。
混合对照品贮备溶液的制备 精密量取上述对照品贮备溶液各1ml,置100ml量瓶中,用异辛烷稀释至刻度,摇匀,即得。
混合对照品溶液的制备 分别精密量取上述混合对照品贮备溶液,用异辛烷制成每1L分别含10μg、20μg、50μg、100μg、200μg、500μg的溶液,即得(其中β-六六六、异狄氏剂、p,p'-滴滴滴、o,p'-滴滴涕每1L分别含20μg、40μg、100μg、200μg、400μg、1000μg)。
供试品溶液的制备 取供试品,粉碎成粉末(过三号筛),取约1.5g,精密称定,置于50ml聚苯乙烯具塞离心管中,加入水10ml,混匀,放置2小时,精密加入乙腈1 5 ml,剧烈振摇提取1分钟,再加入预先称好的无水硫酸镁4g与氯化钠1g的混合粉末,再次剧烈振摇1分钟后,离心(4000r/min)1分钟。精密吸取上清液10ml, 40℃减压浓缩至近干,用环己烷-乙酸乙酯(1∶1)混合溶液分次转移至10ml量瓶中,加环己烷-乙酸乙酯(1∶1)混合溶液至刻度,摇匀,转移至预先加入1g无水硫酸钠的离心管中,振摇,放置1小时,离心(必要时滤过),取上清液5ml过凝胶渗透色谱柱[400mm×25mm,内装BIO-Beads S-X3填料;以环己烷-乙酸乙酯(1∶1)混合溶液为流动相;流速为每分钟5.0ml]净化,收集18~30分钟的洗脱液,于40℃水浴减压浓缩至近干,加少量正己烷替换两次,加正己烷1ml使溶解,转移至弗罗里硅土固相萃取小柱[1000mg/6ml,用正己烷-丙酮(95∶5)混合溶液10ml和正己烷10ml预洗]上,残渣用正己烷洗涤3次,每次1ml,洗液转移至同一弗罗里硅土固相萃取小柱上,再用正己烷-丙酮(95∶5)混合溶液10ml洗脱,收集全部洗脱液,置氮吹仪上吹至近干,加异辛烷定容至1ml,涡旋使溶解,即得。
表1 22种有机氯类农药对照品贮备液浓度、相对保留时间及检出限参考值
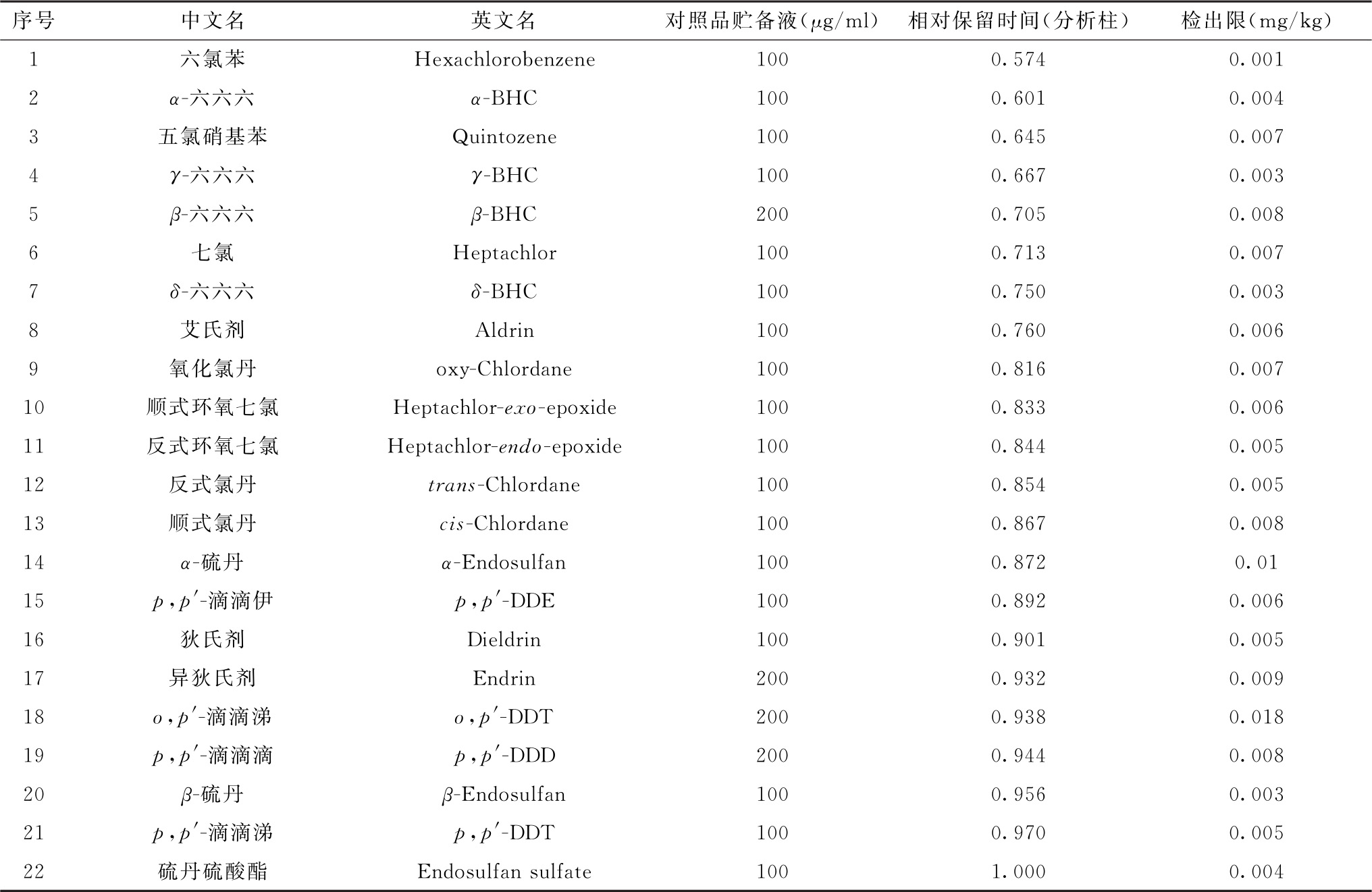
注:各对照品的相对保留时间以硫丹硫酸盐为参照峰计算。
测定法 分别精密吸取供试品溶液和混合对照品溶液各1μl,注入气相色谱仪,按外标标准曲线法计算供试品中22种有机氯农药残留量。
【附注】(1)当供试品中有农药检出时,可在验证柱中确认检出的结果,再进行定量。必要时,可对检出的结果用气相色谱-串联质谱法进行确证。
(2)加样回收率应在70%~120%之间。
第二法 有机磷类农药残留量测定法(色谱法)
色谱条件与系统适用性试验 以50%苯基-50%二甲基聚硅氧烷或(5%苯基)甲基聚硅氧烷为固定液的弹性石英毛细管柱(0.25mm×30m, 0.25μm),氮磷检测器(NPD)或火焰光度检测器(FPD)。进样口温度220℃,检测器温度300℃,不分流进样。程序升温:初始120℃,每分钟10℃升至200℃;每分钟5℃升至240℃,保持2分钟;每分钟20℃升至270℃,保持0.5分钟。理论板数按敌敌畏峰计算应不低于6000,两个相邻色谱峰的分离度应大于1.5。
对照品贮备溶液的制备 精密称取对硫磷、甲基对硫磷、乐果、氧化乐果、甲胺磷、久效磷、二嗪磷、乙硫磷、马拉硫磷、杀扑磷、敌敌畏、乙酰甲胺磷农药对照品适量,用乙酸乙酯分别制成每1ml约含100μg的溶液,即得。
混合对照品贮备溶液的制备 分别精密量取上述各对照品贮备溶液1ml,置20ml棕色量瓶中,加乙酸乙酯稀释至刻度,摇匀,即得。
混合对照品溶液的制备 精密量取上述混合对照品贮备溶液,用乙酸乙酯制成每1ml含0.1μg、0.5μg、1μg、2μg、5μg的浓度系列,即得。
供试品溶液的制备 药材或饮片 取供试品,粉碎成粉末(过三号筛),取约5g,精密称定,加无水硫酸钠5g,加入乙酸乙酯50~100ml,冰浴超声处理3分钟,放置,取上层液滤过,药渣加入乙酸乙酯30~50ml,冰浴超声处理2分钟,放置,滤过,合并两次滤液,用少量乙酸乙酯洗涤滤纸及残渣,与上述滤液合并。取滤液于40℃以下减压浓缩至近干,用乙酸乙酯转移至5ml量瓶中,并稀释至刻度;精密吸取上述溶液1ml,置石墨化炭小柱(250mg/3ml用乙酸乙酯5ml预洗)上,用正己烷-乙酸乙酯(1∶1)混合溶液5ml洗脱,收集洗脱液,置氮吹仪上浓缩至近干,加乙酸乙酯定容至1ml,涡旋使溶解,即得。
测定法 分别精密吸取供试品溶液和与之相对应浓度的混合对照品溶液各1μl,注入气相色谱仪,按外标法计算供试品中12种有机磷农药残留量。
第三法 拟除虫菊酯类农药残留量测定法(色谱法)
色谱条件与系统适用性试验 以(5%苯基)甲基聚硅氧烷为固定液的弹性石英毛细管柱(0.32mm×30m, 0.25μm),63Ni-ECD电子捕获检测器。进样口温度270℃,检测器温度330℃。不分流进样(或根据仪器设置最佳的分流比)。程序升温:初始160℃,保持1分钟;每分钟10℃升至278℃,保持0.5分钟;每分钟1℃升至290℃,保持5分钟。理论板数按溴氰菊酯峰计算应不低于105,两个相邻色谱峰的分离度应大于1.5。
对照品贮备溶液的制备 精密称取氯氰菊酯、氰戊菊酯及溴氰菊酯农药对照品适量,用石油醚(60~90℃)分别制成每1ml含20~25μg的溶液,即得。
混合对照品贮备溶液的制备 精密量取上述各对照品贮备液1ml,置10ml量瓶中,用石油醚(60~90℃)稀释至刻度,摇匀,即得。
混合对照品溶液的制备 精密量取上述混合对照品贮备液,用石油醚(60~90℃)制成每1L分别含0、2μg、8μg、40μg、200μg的溶液,即得。
供试品溶液的制备 药材或饮片 取供试品,粉碎成粉末(过三号筛),取1~2g,精密称定,置100ml具塞锥形瓶中,加石油醚(60~90℃)-丙酮(4∶1)混合溶液30ml,超声处理15分钟,滤过,药渣再重复上述操作2次后,合并滤液,滤液用适量无水硫酸钠脱水后,于40~45℃减压浓缩至近干,用少量石油醚(60~90℃)反复操作至丙酮除净,残渣用适量石油醚(60~90℃)溶解,置混合小柱[从上至下依次为无水硫酸钠2g、弗罗里硅土4g、微晶纤维素1g、氧化铝1g、无水硫酸钠2g,用石油醚(60~90℃)-乙醚(4∶1)混合溶液20ml预洗]上,用石油醚(60~90℃)-乙醚(4∶1)混合溶液90ml洗脱,收集洗脱液,于40~45℃减压浓缩至近干,再用石油醚(60~90℃)3~4ml重复操作至乙醚除净,用石油醚(60~90℃)溶解并转移至5ml量瓶中,并稀释至刻度,摇匀,即得。
测定法 分别精密吸取供试品溶液和与之相对应浓度的混合对照品溶液各1μl,注入气相色谱仪,按外标法计算供试品中3种拟除虫菊酯农药残留量。
第四法 农药多残留量测定法(质谱法)
一、定性测定方法
本法系用气相色谱-串联质谱法与液相色谱-串联质谱法对中药中农药残留的快速定性筛查,发现残留农药,便于农药定量测定。实验室应建立必要的质控手段,保证定性结果准确。
1 气相色谱-串联质谱法
色谱条件 以(5%苯基)甲基聚硅氧烷为固定液的弹性石英毛细管柱(0.25mm×30m, 0.25μm)。进样口温度240℃,不分流进样。载气为高纯氦气(He)。进样口为恒压模式,柱前压力为146kPa(可依据表2中农药保留时间调整柱前压力)。程序升温:初始温度70℃,保持2分钟,先以每分钟25℃升至150℃,再以每分钟3℃升至200℃,最后以每分钟8℃升至280℃,保持10分钟。
质谱条件 以三重四极杆串联质谱仪检测;离子源为电子轰击源(EI),离子源温度280℃。碰撞气为氮气或氩气,流速1.5ml/min。质谱传输接口温度280℃。质谱监测模式为多反应监测(MRM),各化合物参考保留时间、监测离子对、碰撞电压(CE)与检出限参考值见表2。为提高检测灵敏度,可根据保留时间分段监测各农药。
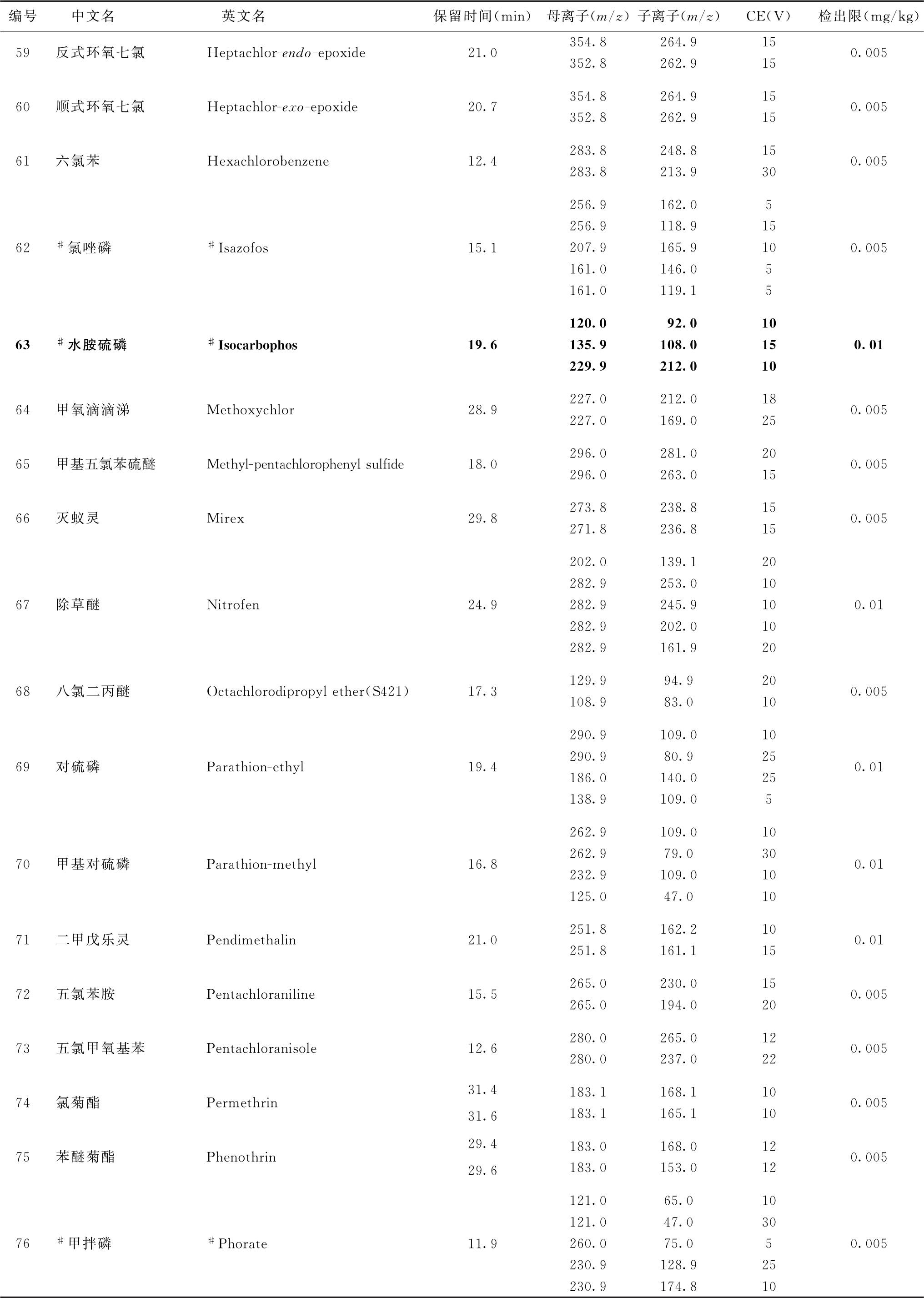
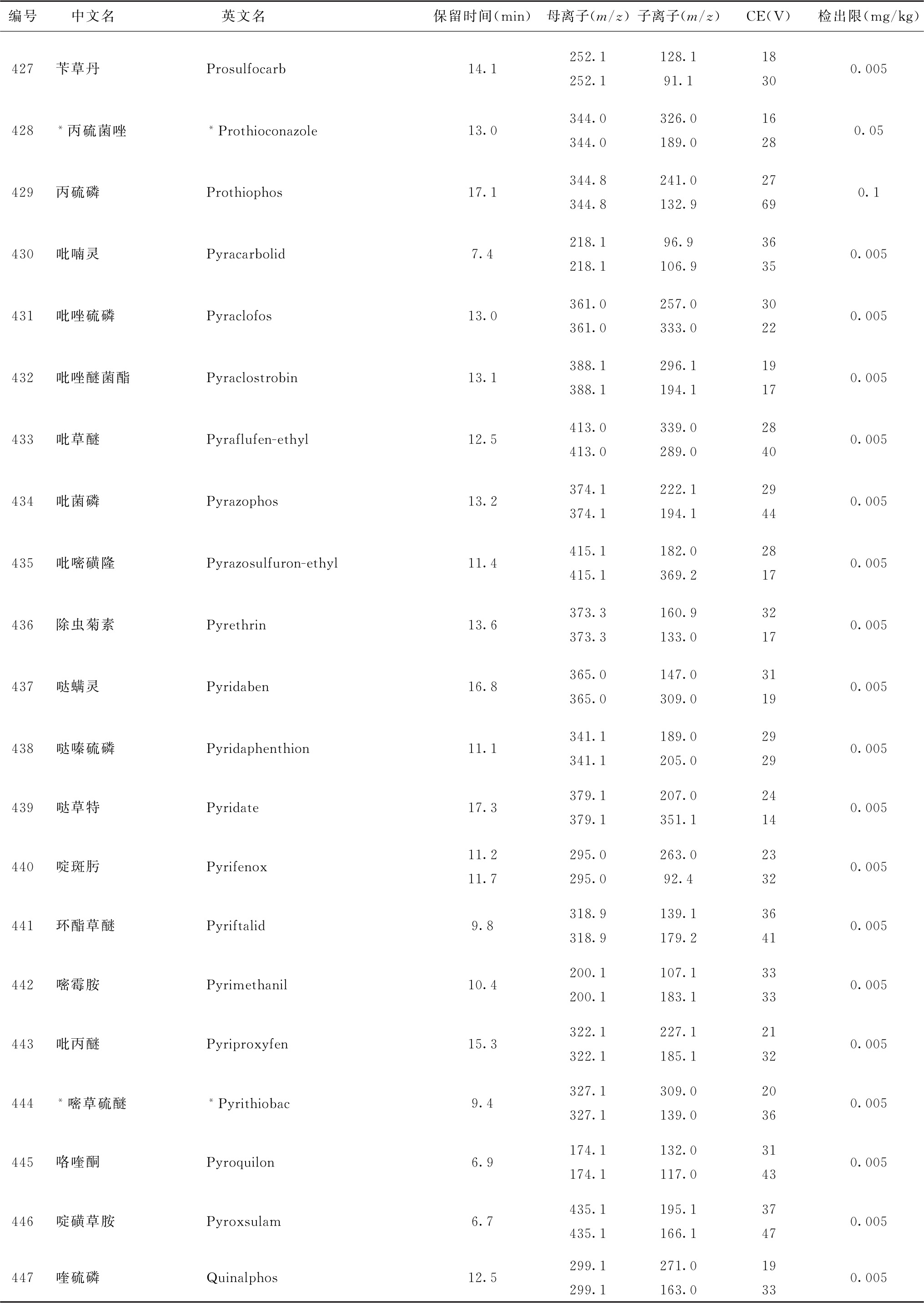
表2 91种农药及内标对照品的保留时间、监测离子对、碰撞电压(CE)与检出限参考值
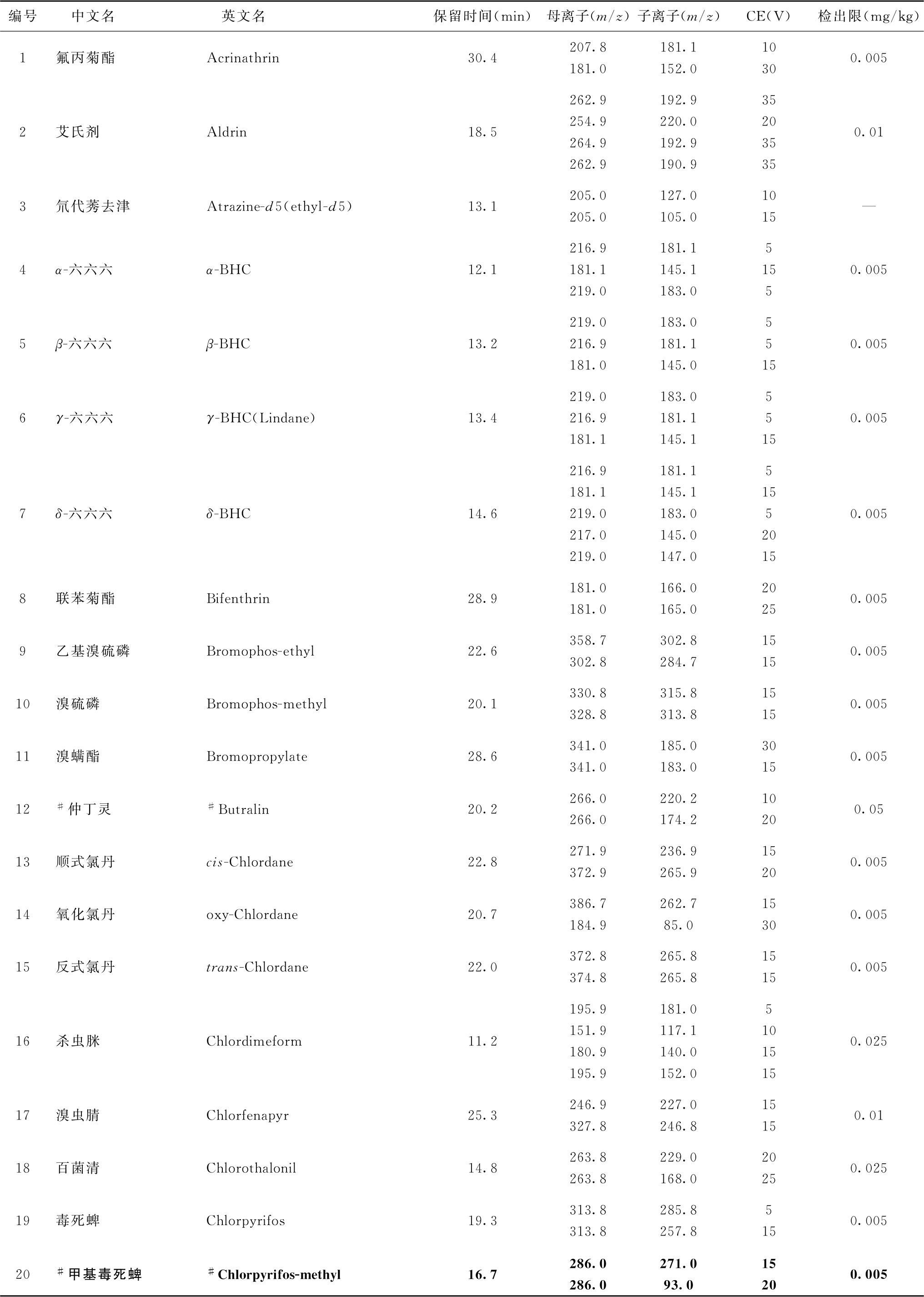
续表

续表
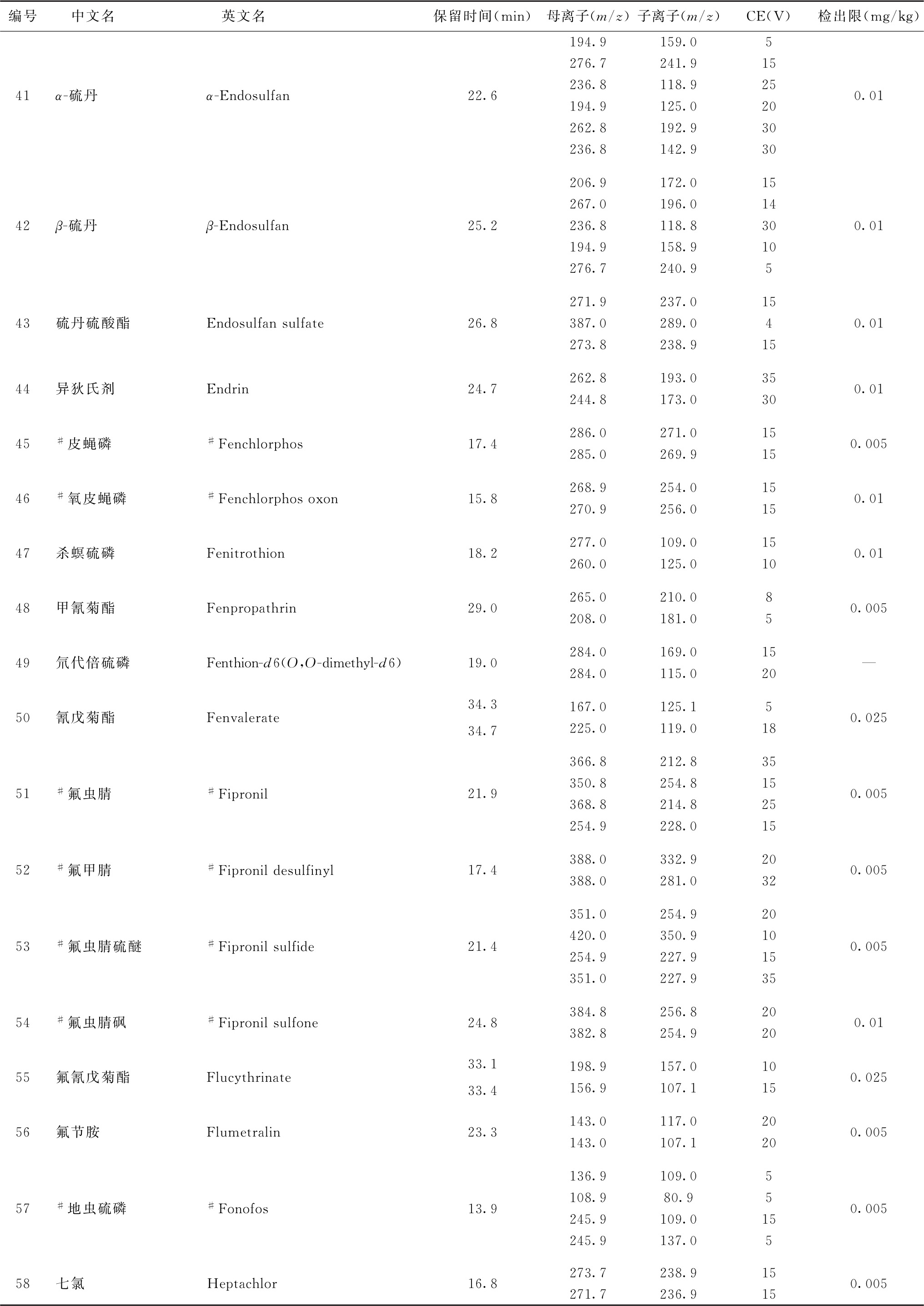
续表
续表
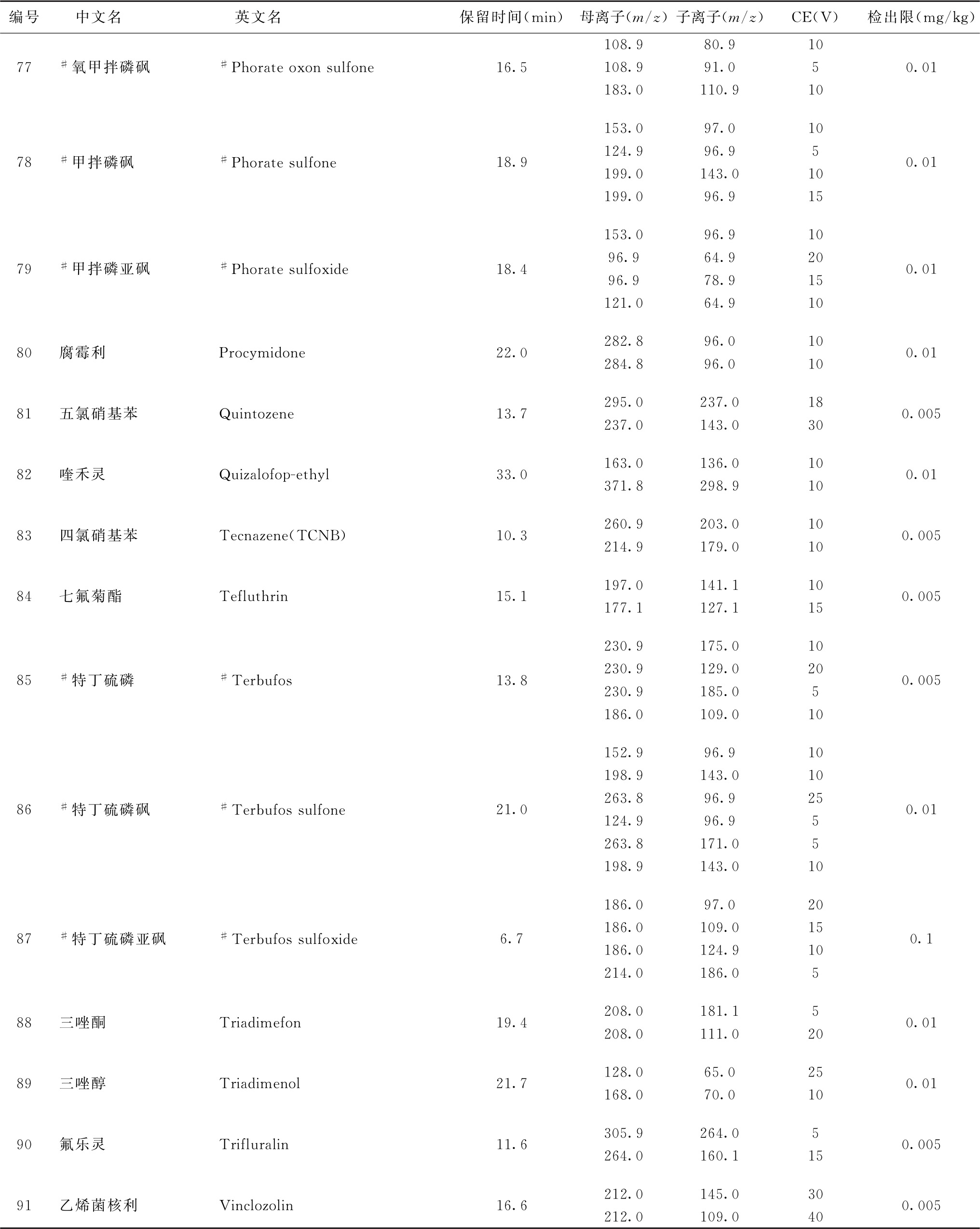
注:1.表中化合物3、32与49为内标。
2.部分化合物含有多个异构体,表中提供了多个异构体峰的参考保留时间。
3.鉴于中药材基质的复杂性,为便于方法使用,部分化合物提供了多个监测离子对。依情况选择2个监测离子对用于测定。
4.标记“#”的化合物为气相色谱-串联质谱法与液相色谱-串联质谱法均有定性定量方法的化合物,一般情况下优先选择在表2或表4中字体被加粗的化合物所对应的测定方法,测定存在干扰时可选用另一测定方法。
定性用混合对照品溶液的制备 取本法“二、定量测定方法”中的基质混合对照品溶液作为定性用混合对照品溶液,即得。
供试品溶液的制备 取本法“二、定量测定方法”中的供试品溶液,即得。
测定法 分别精密吸取供试品溶液和定性用混合对照品溶液各1μl,注入气相色谱-串联质谱仪,按保留时间与定性离子相对丰度比对88种农药进行定性测定。
结果判断 供试品色谱中如检出与对照品保留时间相同的色谱峰,并且在扣除背景后的质谱图中,所选择的2对监测离子对均出现,供试品溶液的监测离子对峰面积比与浓度相当的对照品溶液的监测离子对峰面积比进行比较时,相对偏差不超过下列规定的范围,则可判定样品中存在该农药:相对比例>50%,允许±20%偏差;相对比例20%~50%,允许±25%偏差;相对比例10%~20%,允许±30%偏差;相对比例<10%,允许±50%偏差。
2 液相色谱-串联质谱法
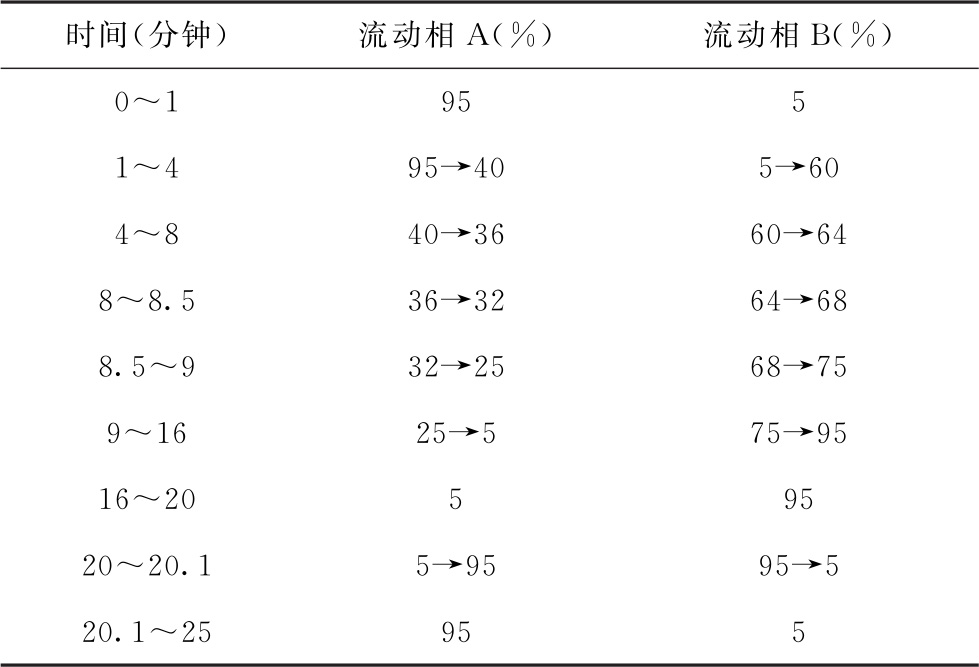
色谱条件 以十八烷基硅烷键合核壳硅胶为填充剂(3mm×150mm, 2.7μm);以0.05%甲酸溶液(含10mmol/L甲酸铵)为流动相A,以0.05%甲酸的甲醇溶液(含10mmol/L甲酸铵)为流动相B,按表3进行梯度洗脱;柱温为35℃,流速为0.4ml/min。
表3 流动相梯度
质谱条件 以三重四极杆串联质谱仪检测;离子源为电喷雾(ESI)离子源,依据表4选择正离子或负离子扫描模式。监测模式为多反应监测(MRM),各化合物保留时间、监测离子对、碰撞电压(CE)和检出限的参考值见表4。为提高检测灵敏度,可根据保留时间分段监测各农药。
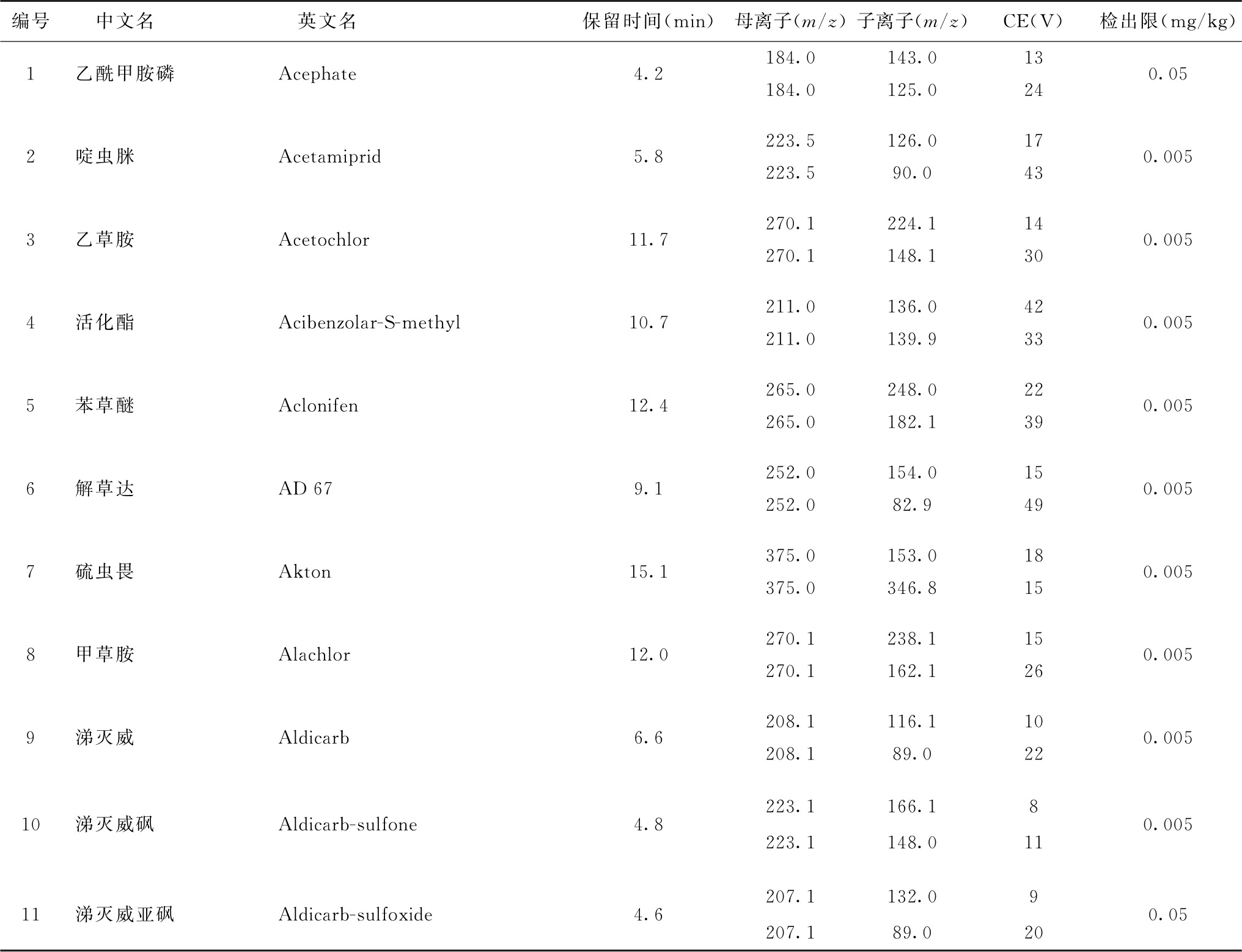
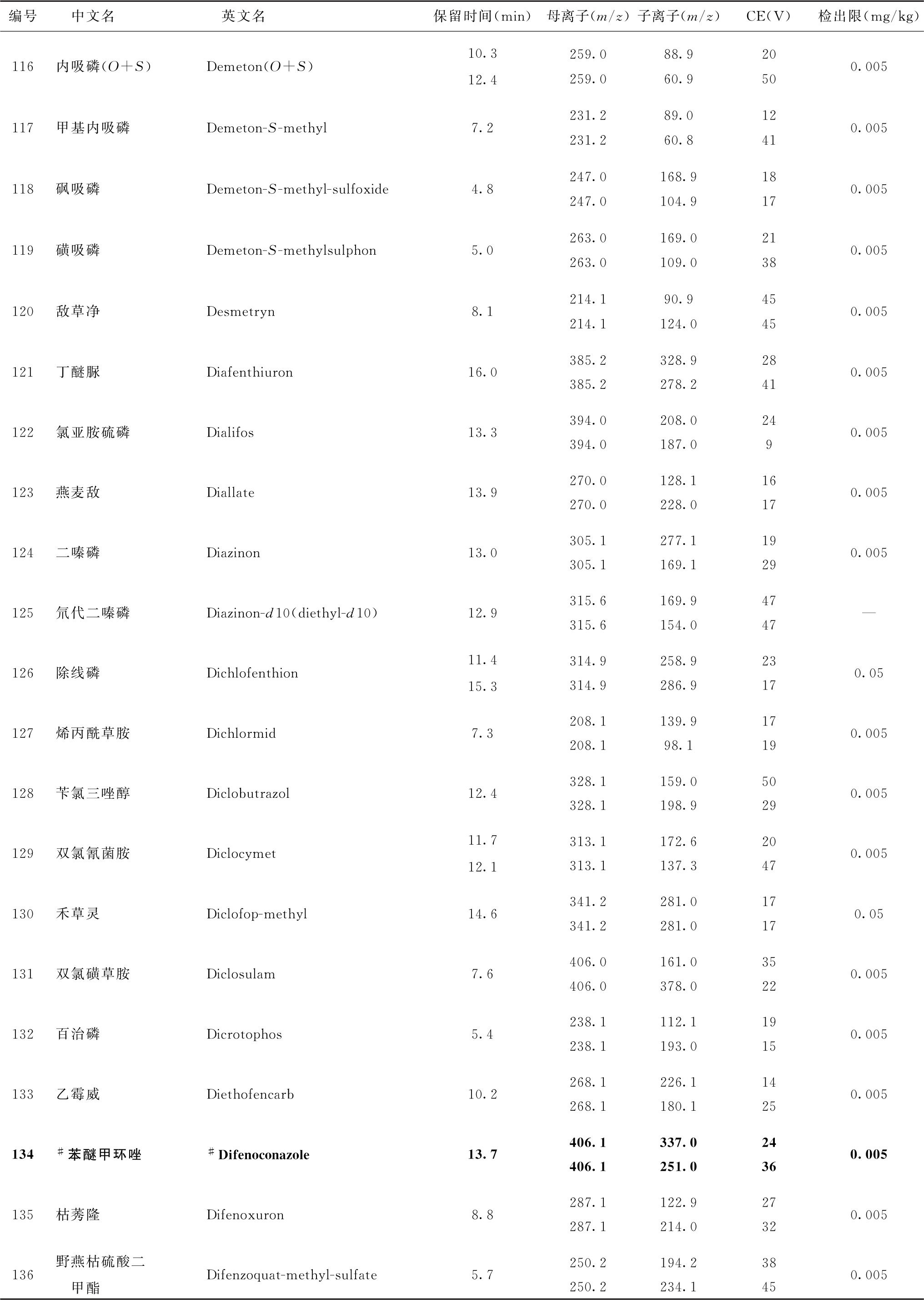
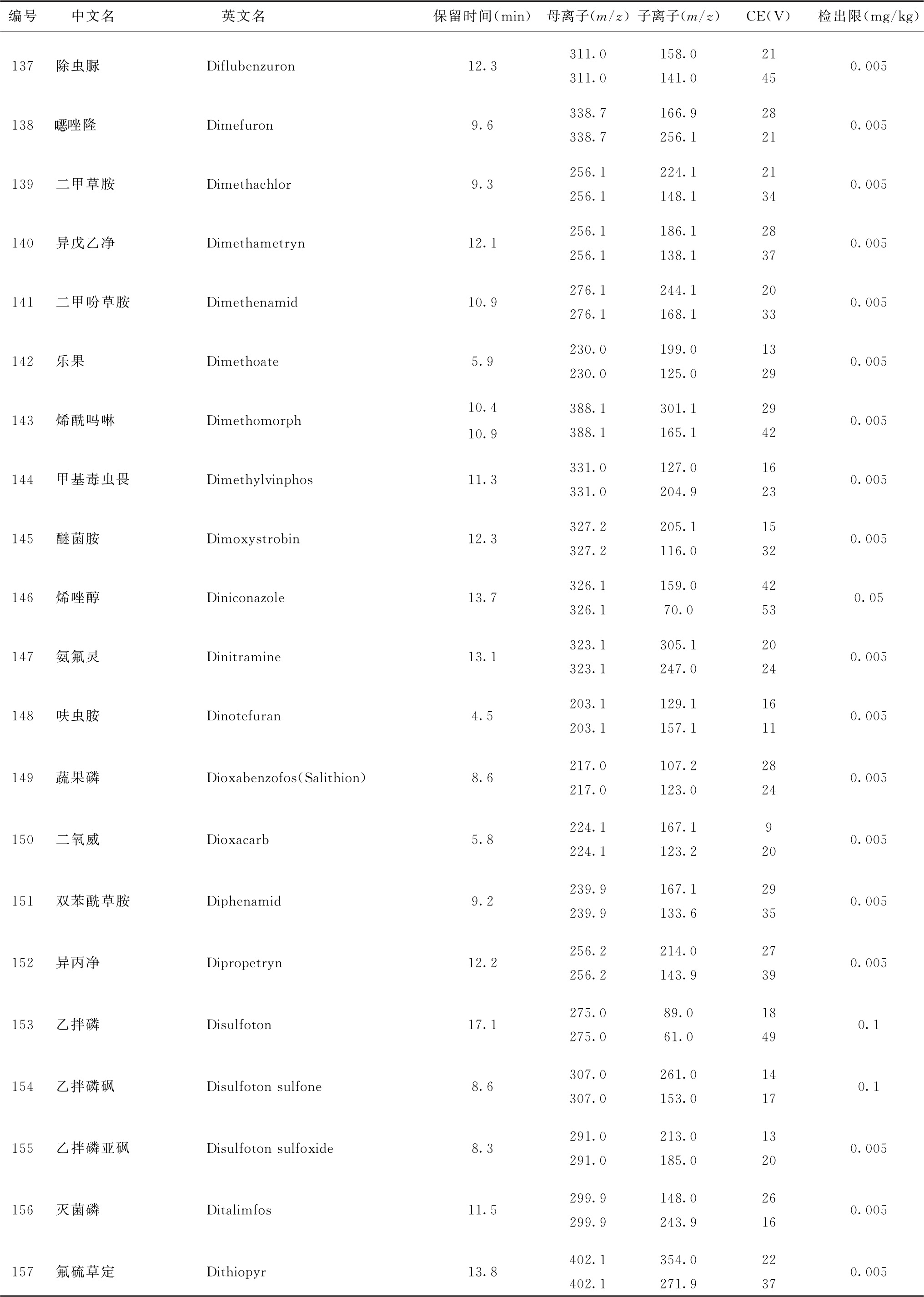
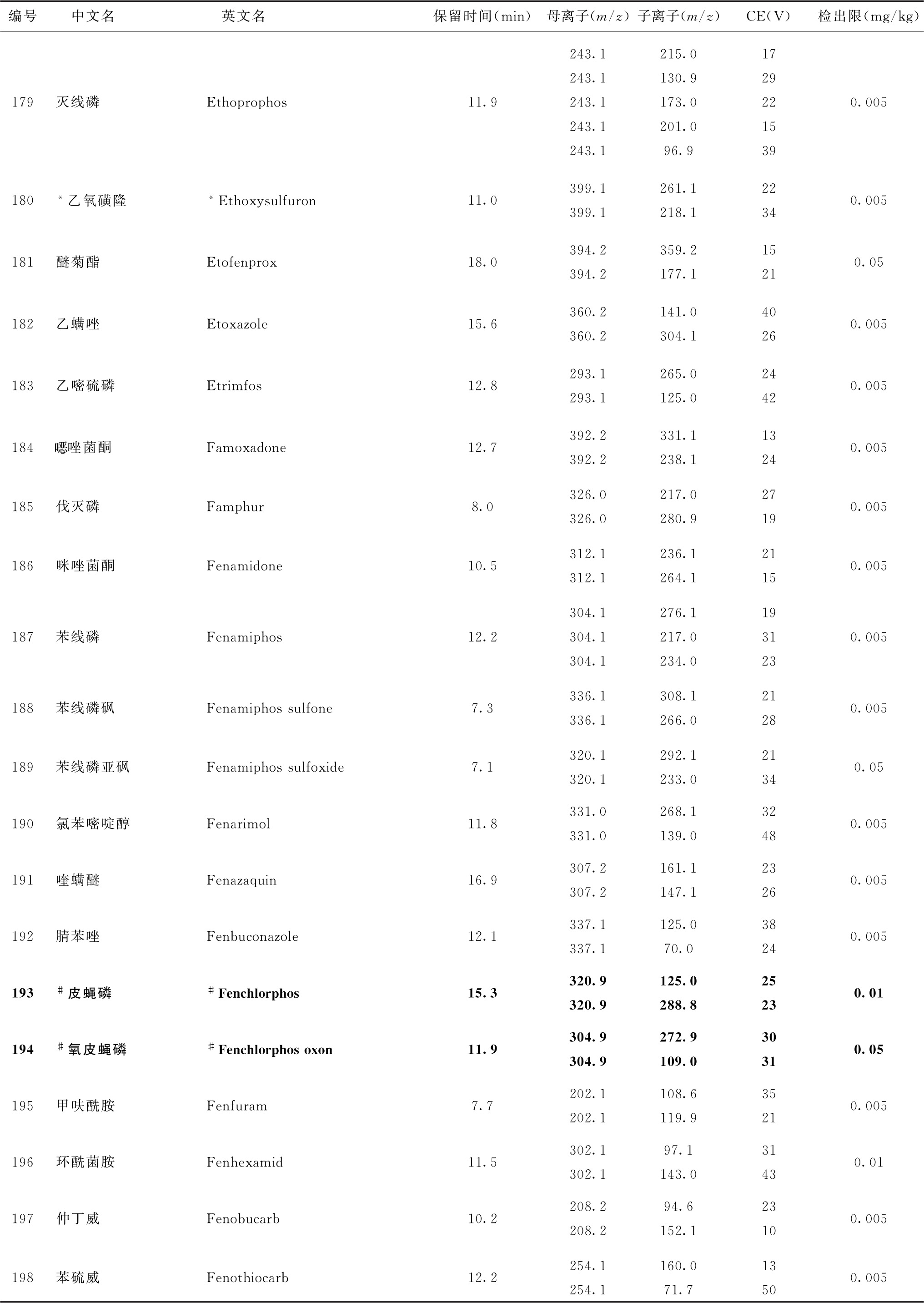
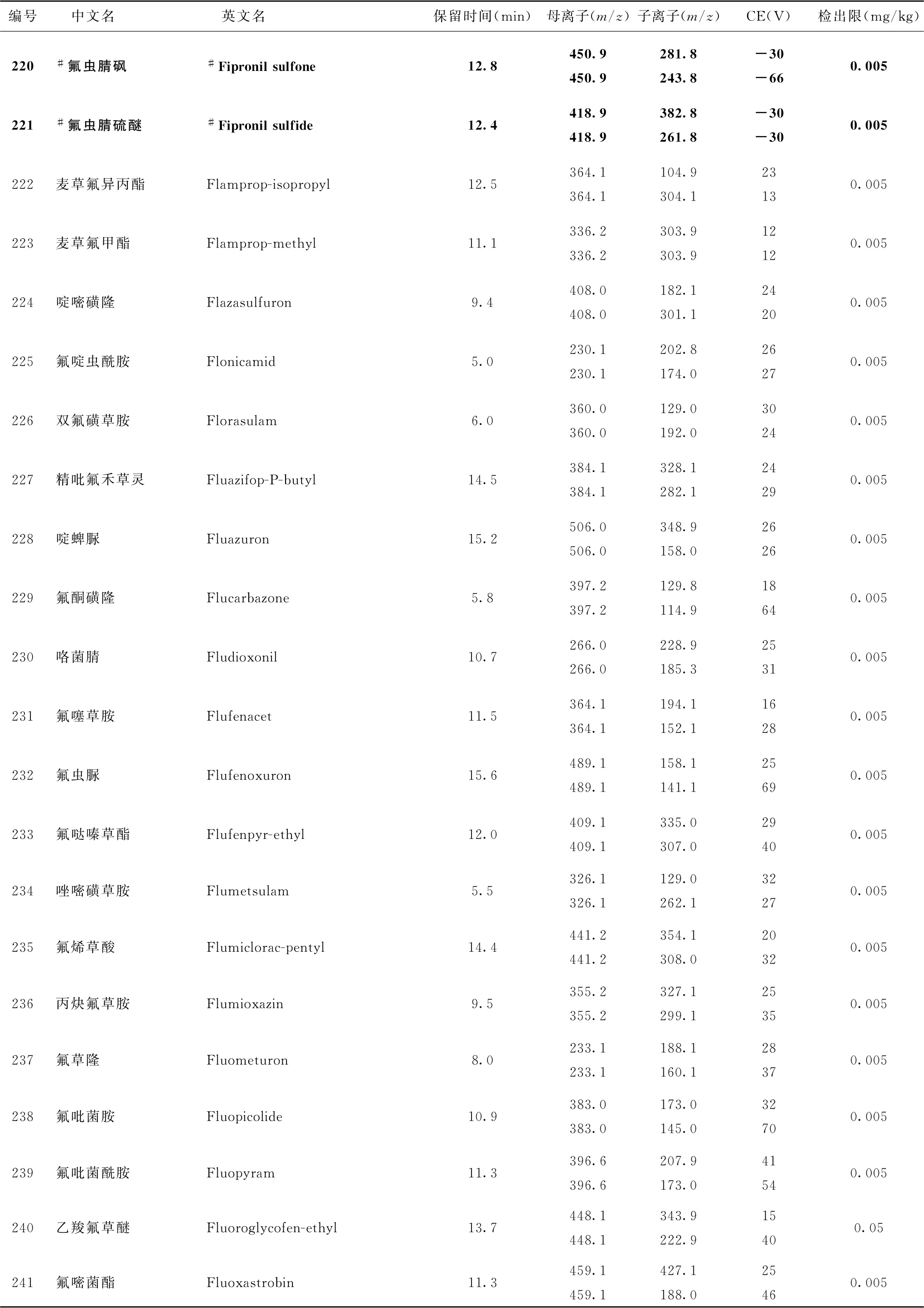
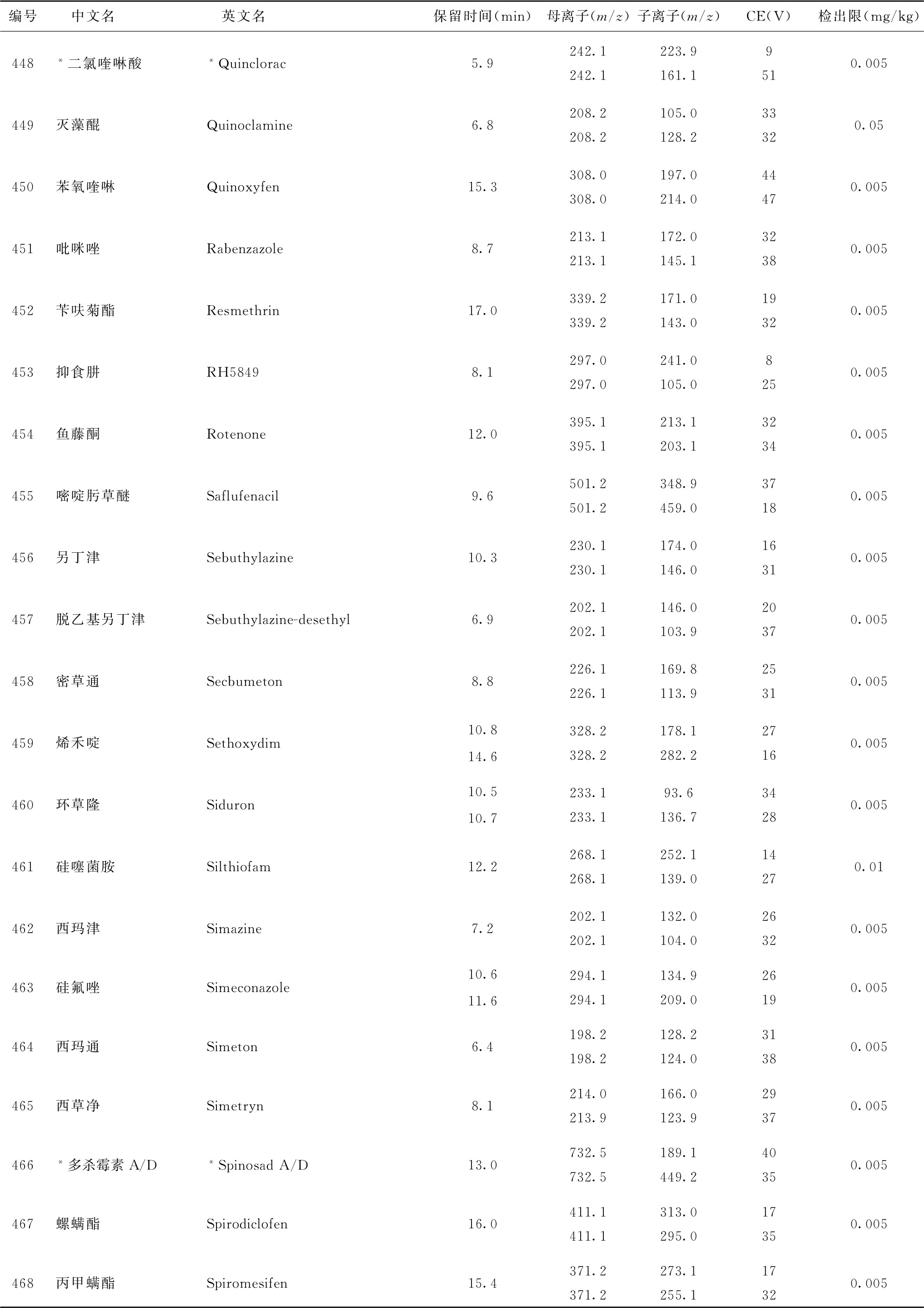
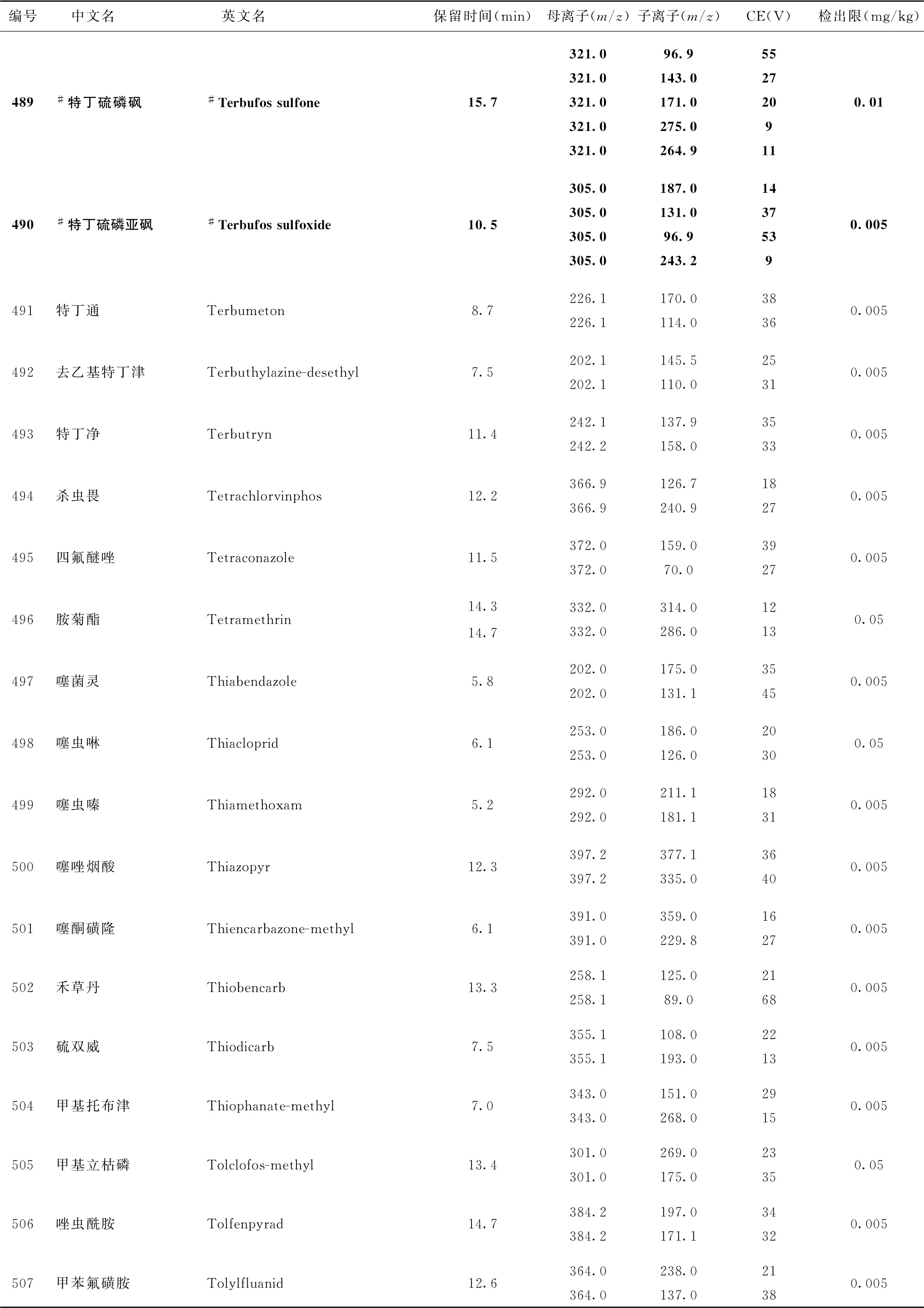
表4 526种农药及内标对照品的保留时间、监测离子对、碰撞电压(CE)与检出限参考值
续表
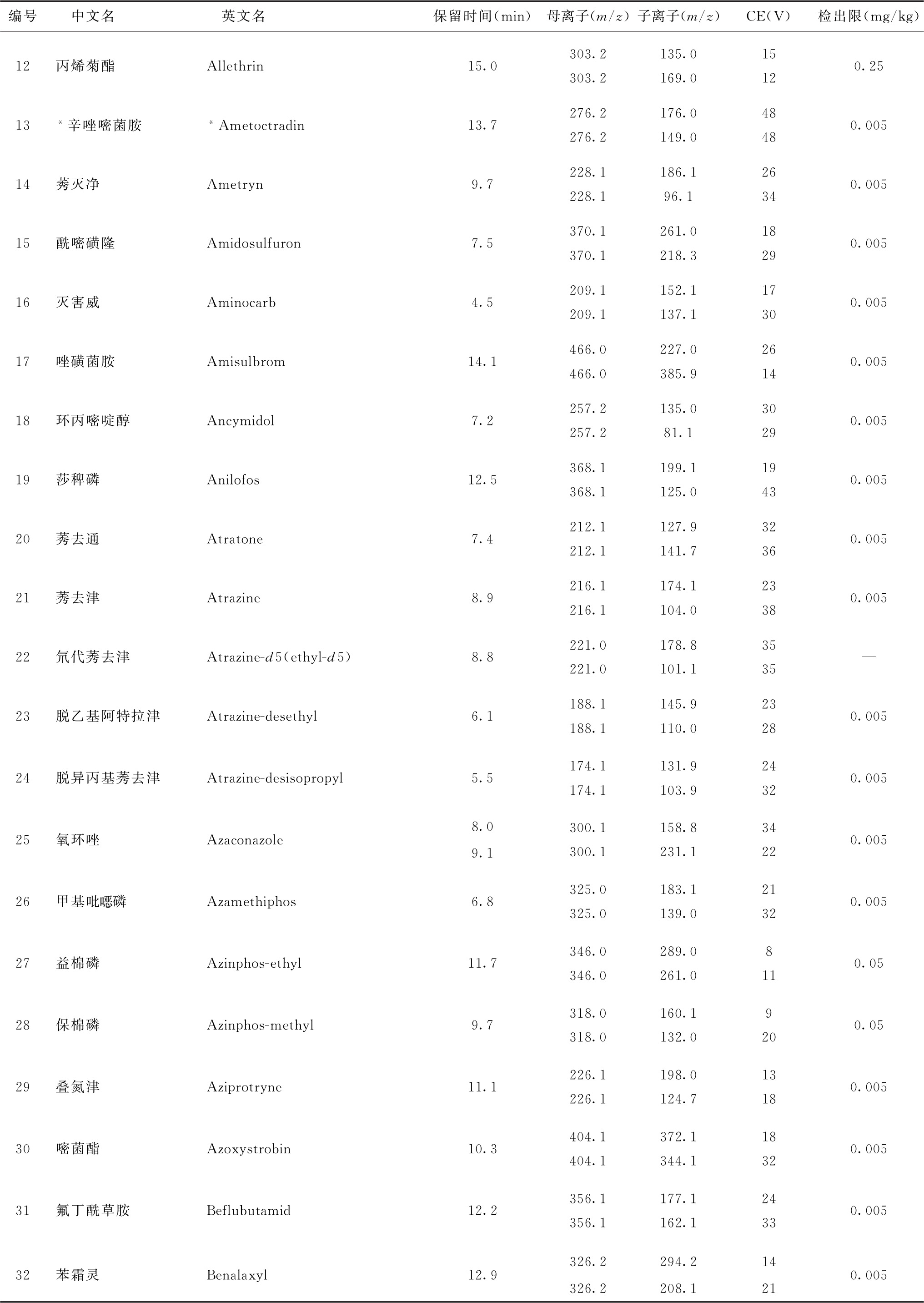
续表
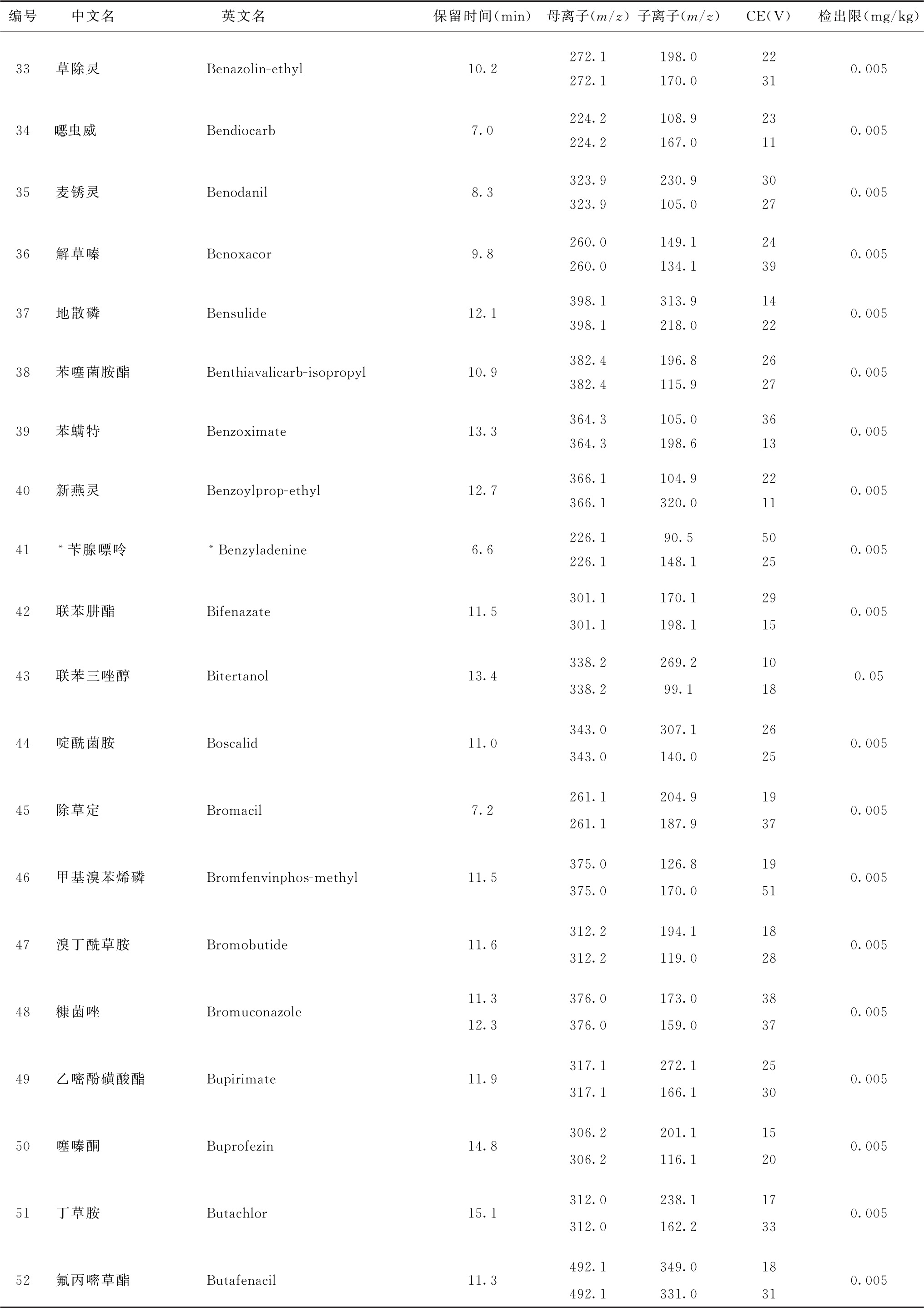
续表
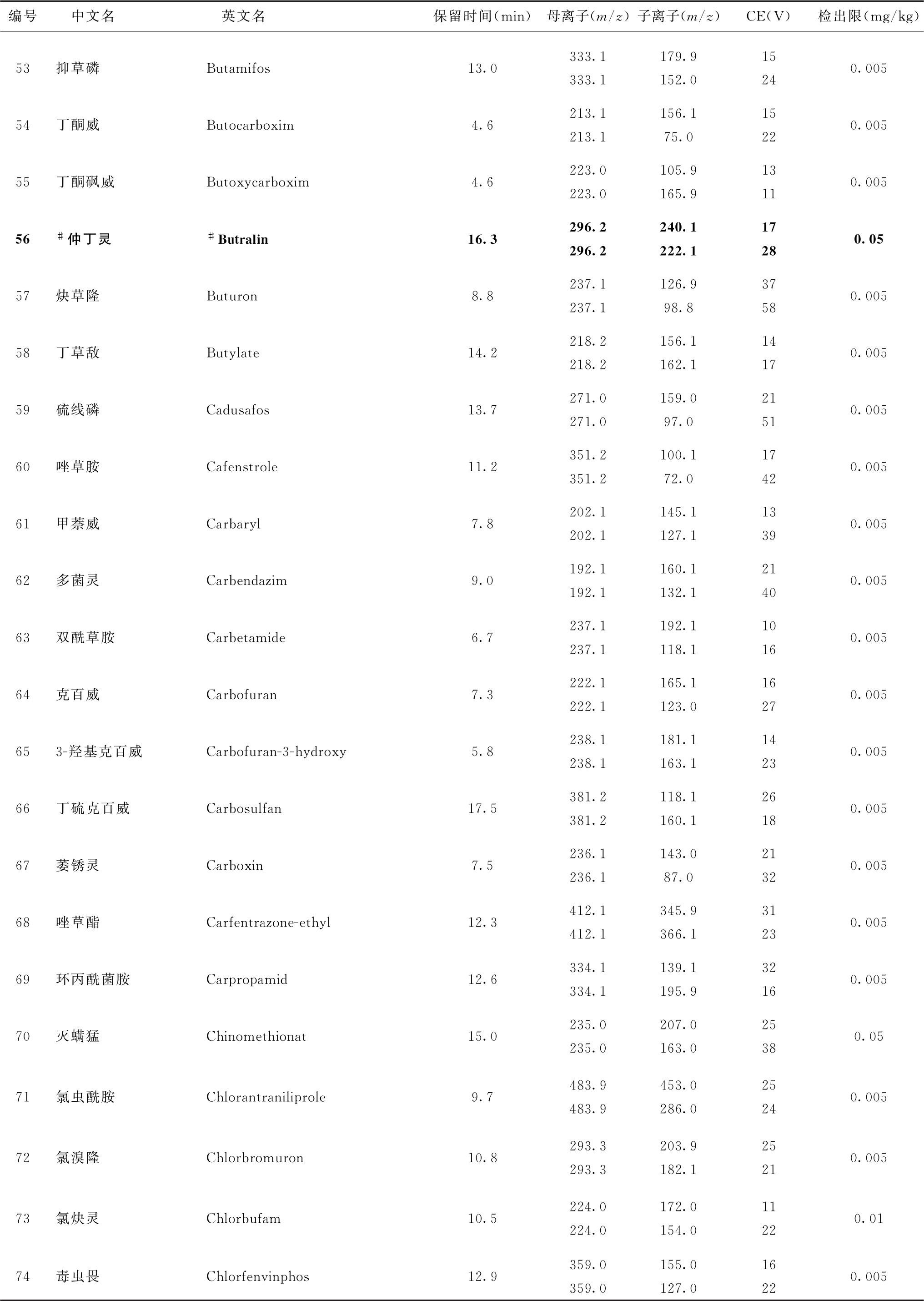
续表

续表
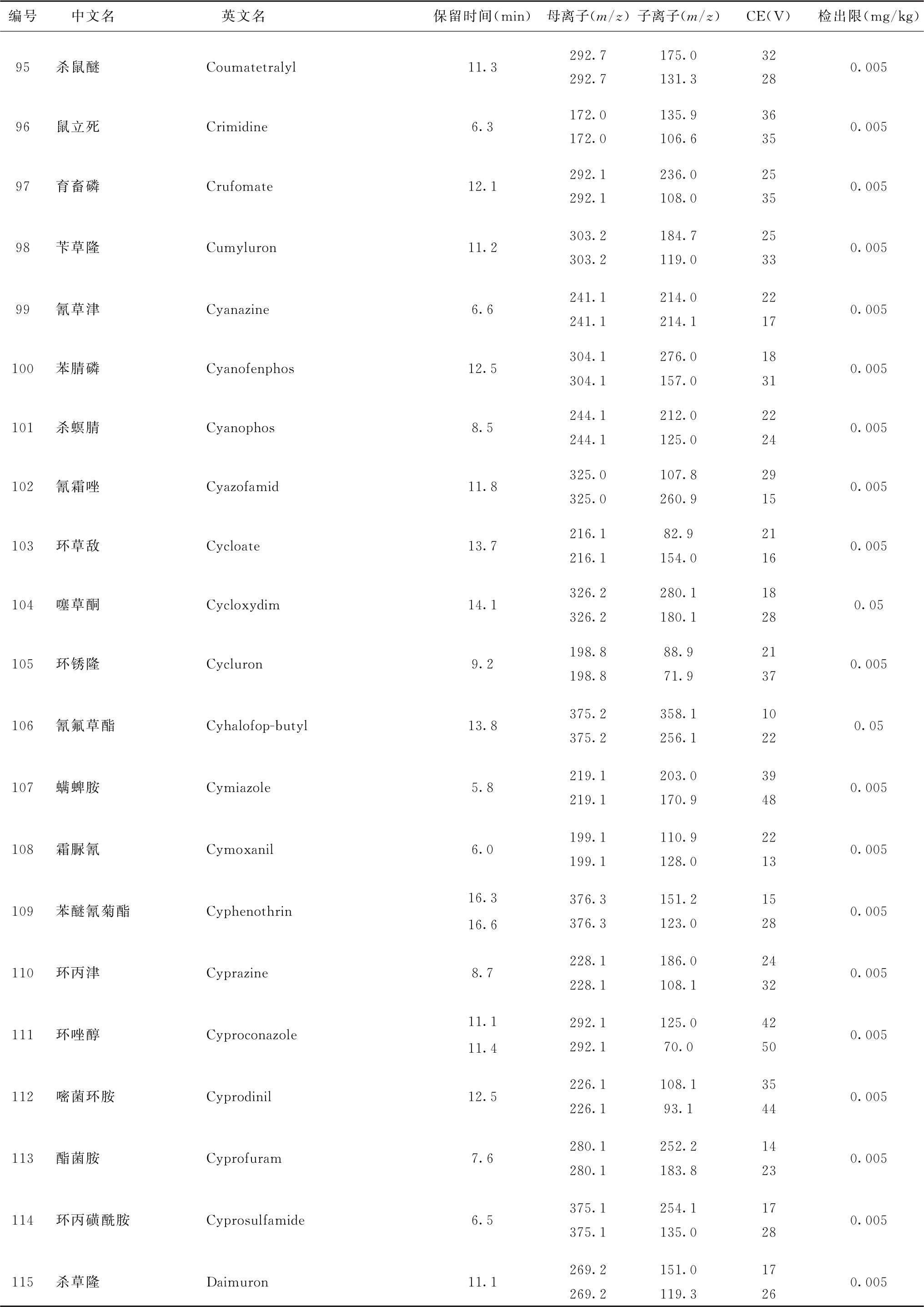
续表
续表
续表

续表
续表

续表
续表
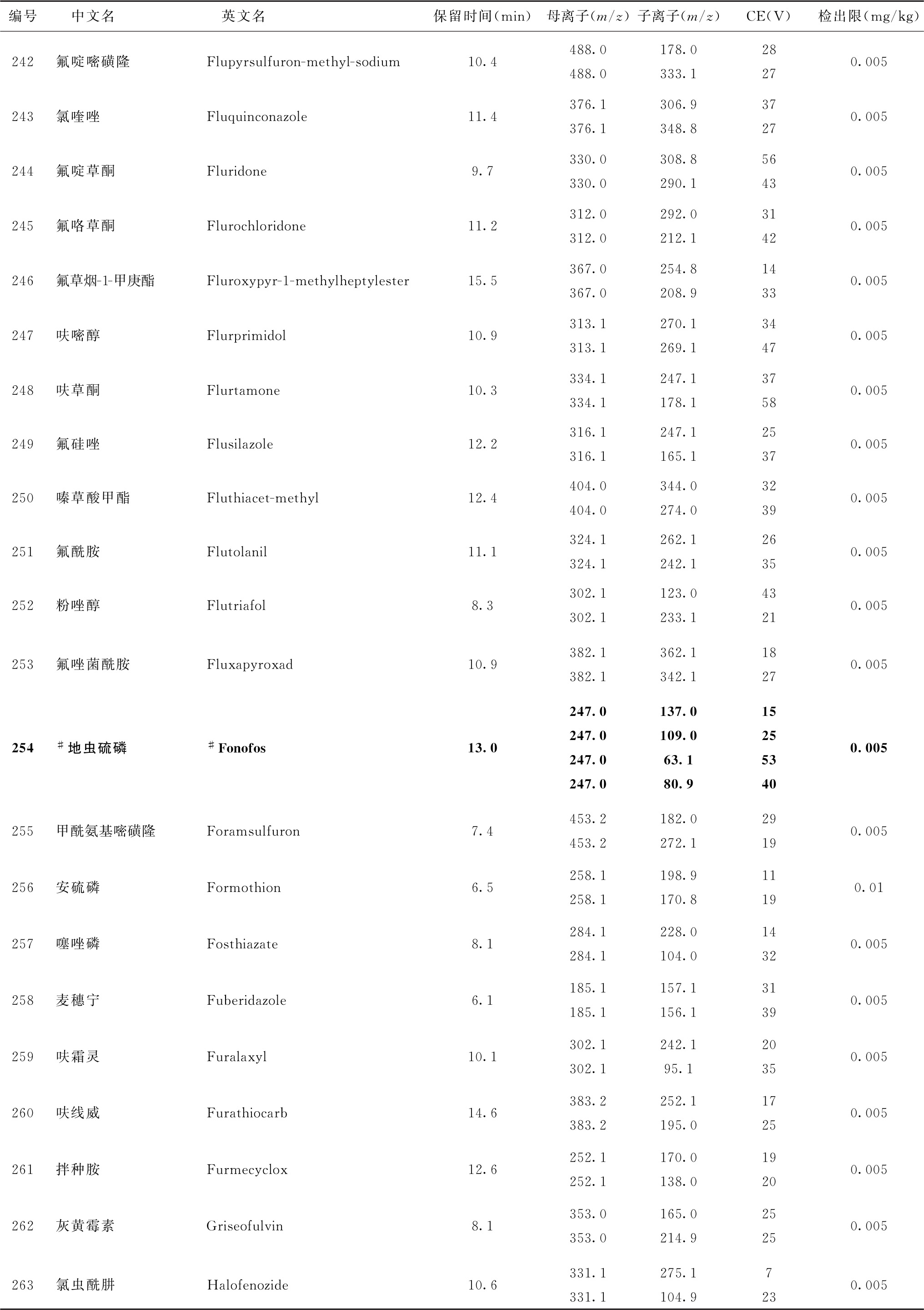
续表
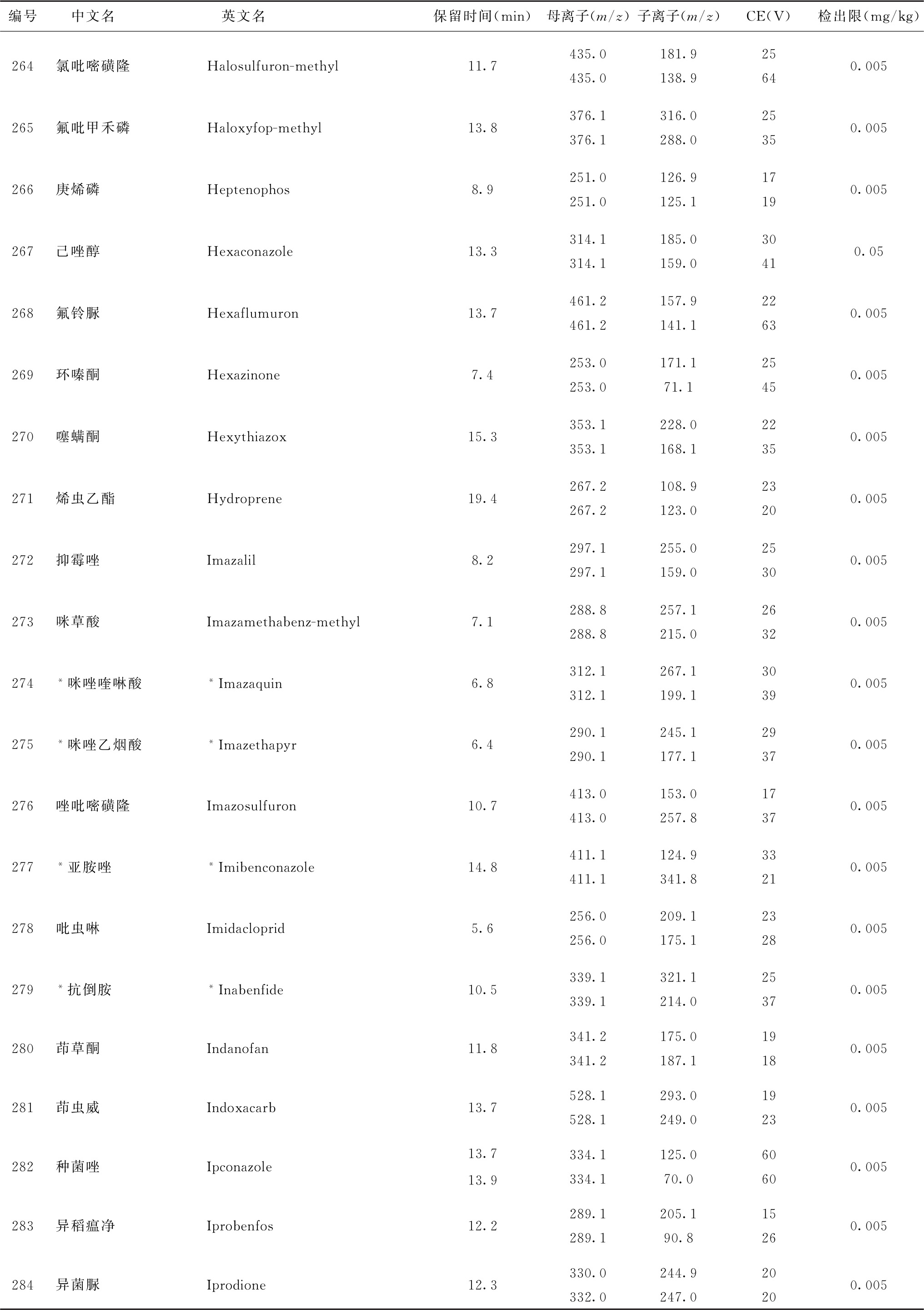
续表
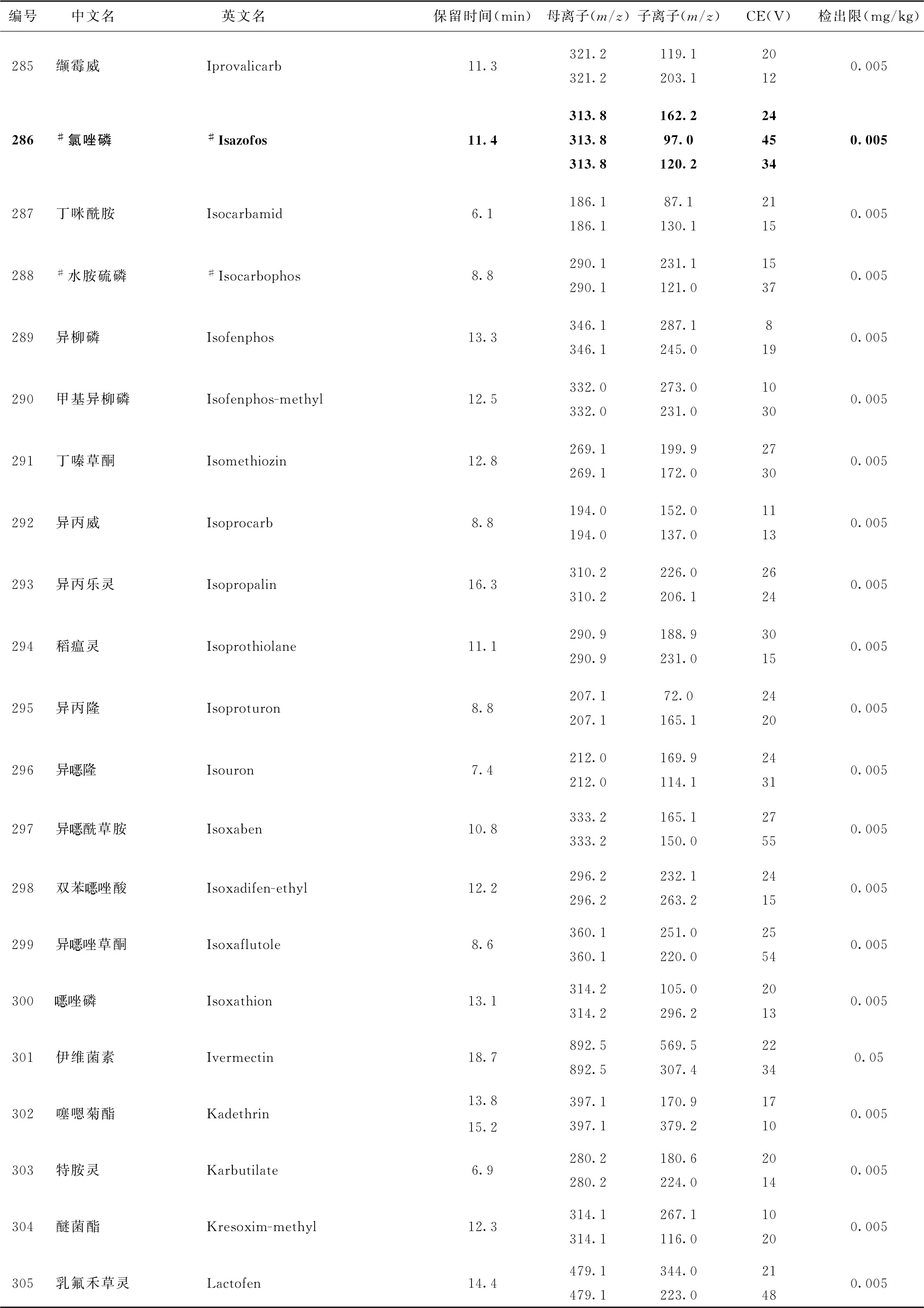
续表

续表
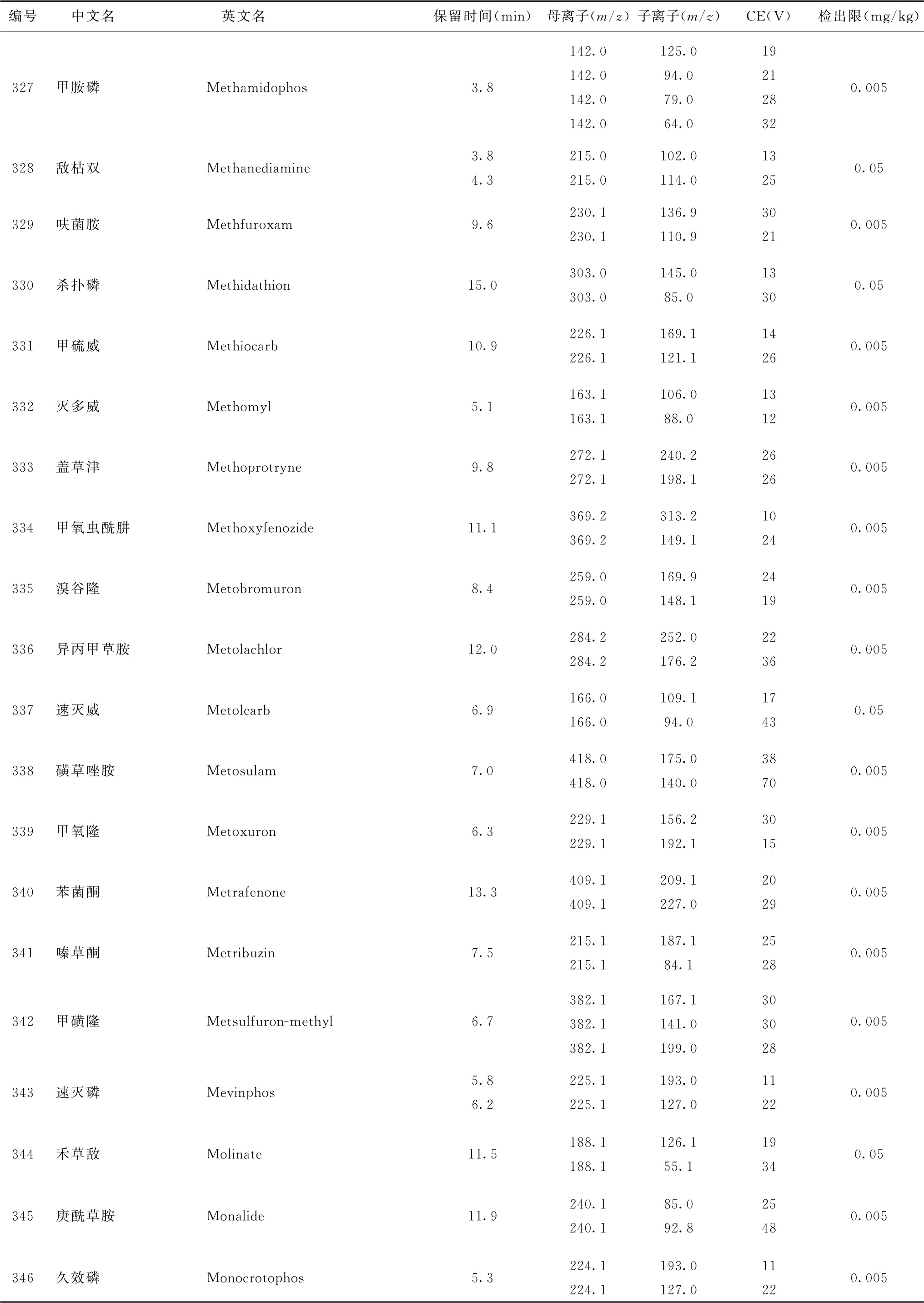
续表
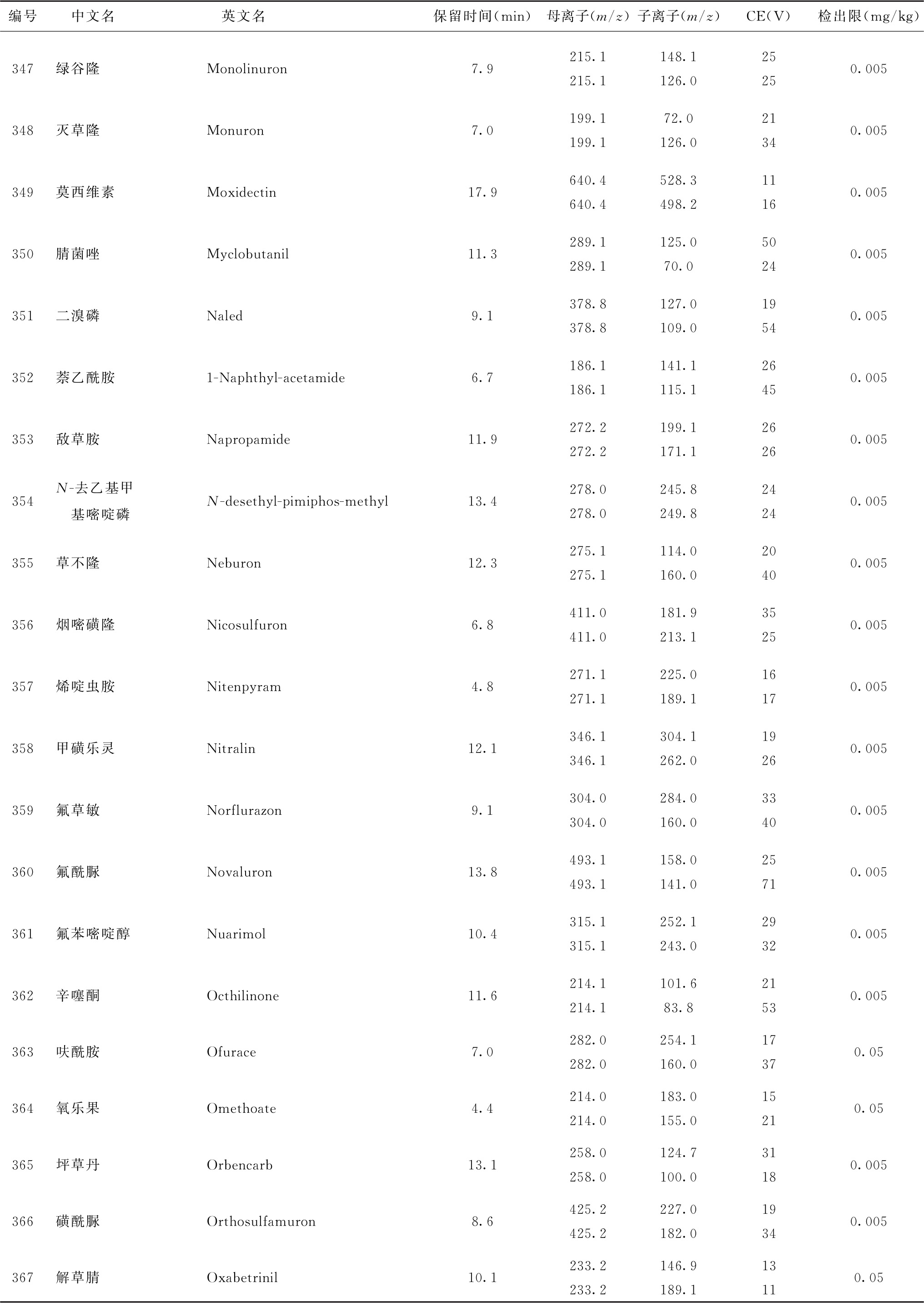
续表
续表
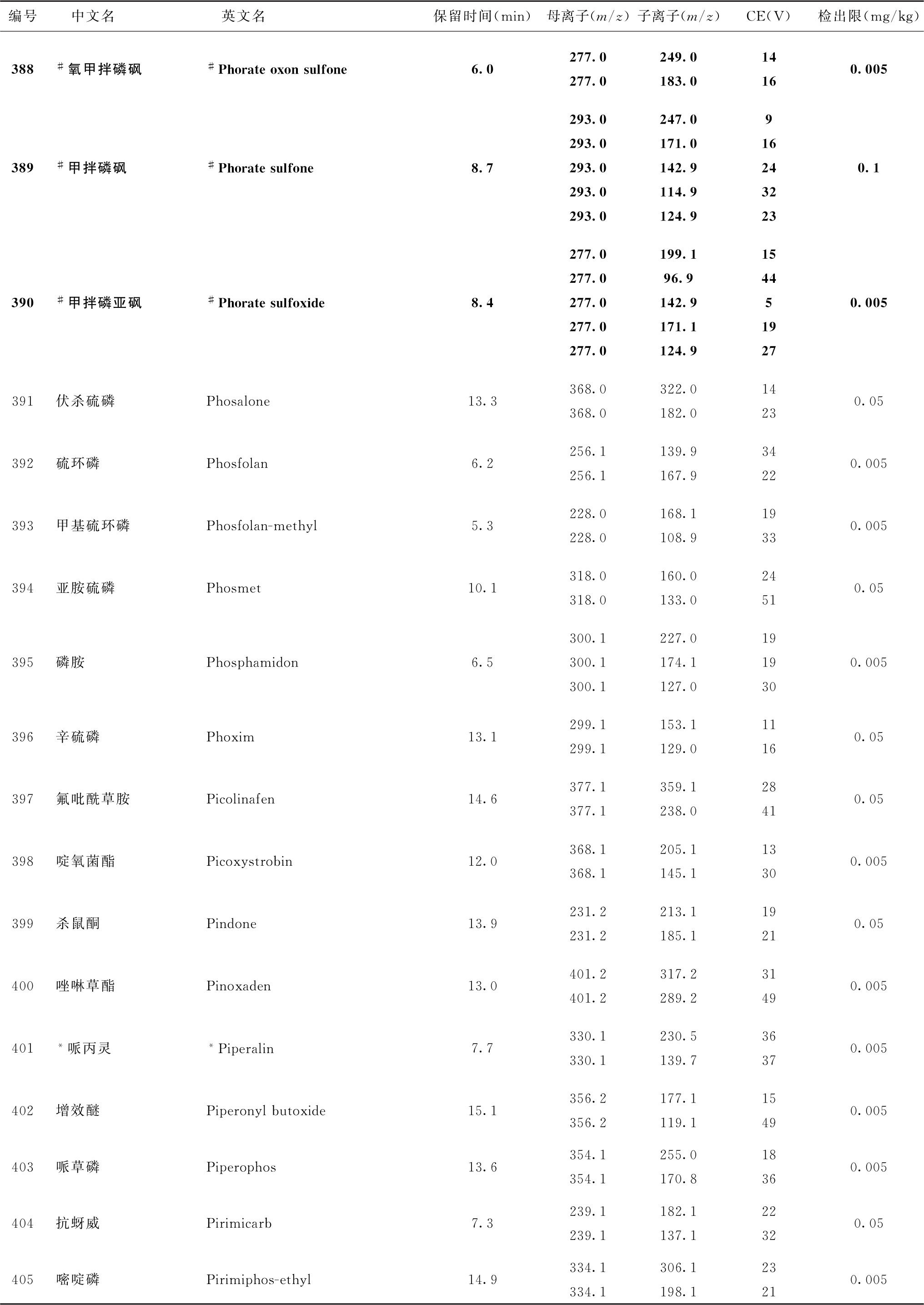
续表
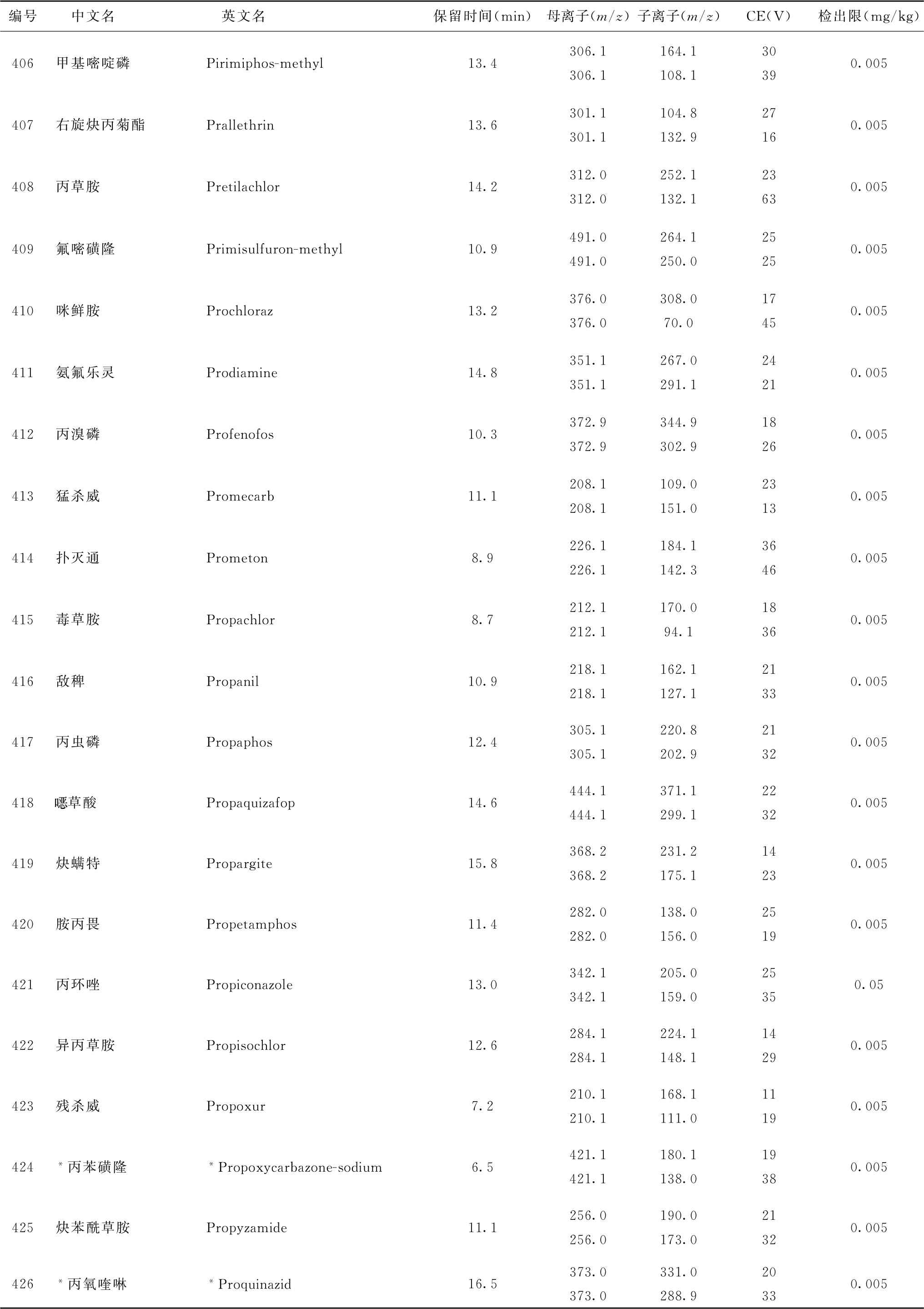
续表
续表
续表

续表
续表

注:1.表中化合物22、125与210为内标。
2.部分化合物含有多个异构体,表中提供了多个异构体峰的参考保留时间。
3.表中218~221化合物使用负离子扫描模式测定,其余化合物采用正离子扫描模式测定。
4.鉴于中药材基质的复杂性,为便于方法使用,部分化合物提供了多个监测离子对。依情况选择2个监测离子对用于测定。
5.标记“*”的化合物使用本法定量测定时回收率低于60%,适用于该化合物定性测定或依【附注】(3)推荐的方法定量测定。
6.标记“#”的化合物为气相色谱-串联质谱法与液相色谱-串联质谱法均有定性定量方法的化合物。一般情况下优先选择在表2或表4中字体被加粗的化合物所对应的测定方法,测定存在干扰时可选用另一测定方法。
定性用混合对照品溶液的制备与供试品溶液的制备 均同本法“一、定性测定方法”中气相色谱-串联质谱法项下。
测定法 分别精密吸取供试品溶液和定性用混合对照品溶液各1~10μl(根据检测要求与仪器灵敏度可适当调整进样量),注入高效液相色谱-串联质谱仪,按保留时间与定性离子相对丰度比对523种农药残留量进行定性测定。
结果判断 供试品色谱中如检出与对照品保留时间相同的色谱峰,并且在扣除背景后的质谱图中,所选择的2对监测离子对均出现,供试品溶液的监测离子对峰面积比与浓度相当的对照品溶液的监测离子对峰面积比进行比较时,相对偏差不超过下列规定的范围,则可判定样品中存在该农药:相对比例>50%,允许±20%偏差;相对比例20%~50%,允许±25%偏差;相对比例10%~20%,允许±30%偏差;相对比例<10%,允许±50%偏差。
二、定量测定方法
本法系用气相色谱-串联质谱法与液相色谱-串联质谱法对中药中农药残留的定量测定方法。实验室应建立必要的质控手段,保证定量结果准确。
1 气相色谱-串联质谱法
色谱条件与质谱条件 均同本法“一、定性测定方法”中气相色谱-串联质谱法项下。
对照品贮备溶液的制备 精密称取表2与表4中农药对照品适量,根据各农药溶解性加乙腈或甲苯分别制成每1ml含1000μg的溶液,即得(可根据具体农药的灵敏度适当调整贮备液配制的浓度)。
内标贮备溶液的制备 取氘代莠去津、氘代二嗪农和氘代倍硫磷对照品适量,精密称定,加乙腈溶解并制成每1ml各含1000μg的混合溶液,即得。
混合对照品溶液的制备 精密量取上述各对照品贮备液适量,用含0.05%醋酸的乙腈分别制成每1L含100μg和1000μg的两种溶液,即得。
内标溶液的制备 精密量取内标贮备溶液适量,加乙腈制成每1ml含6μg的溶液,即得。
基质混合对照品工作溶液的制备 取空白基质样品3g,一式6份,同供试品溶液的制备方法处理至“置氮吹仪上于40℃水浴浓缩至约0.4ml”,分别加入混合对照品溶液(100μg/L)50μl、100μl,混合对照品溶液(1000μg/L)50μl、100μl、200μl、400μl,加乙腈定容至1ml,涡旋混匀,用微孔滤膜滤过(0.22μm),取续滤液,即得浓度分别为5μg/L、10μg/L、50μg/L、100μg/L、200μg/L与400μg/L的系列基质混合对照品溶液。
供试品溶液的制备 药材或饮片 取供试品,粉碎成粉末(过三号筛),取约3g,精密称定,置50ml聚苯乙烯具塞离心管中,加入1%冰醋酸溶液15ml,涡旋使药粉充分浸润,放置30分钟,精密加入乙腈15ml与内标溶液100μl,涡旋使混匀,置振荡器上剧烈振荡(500次/分)5分钟,加入无水硫酸镁与无水乙酸钠的混合粉末(4∶1)7.5g,立即摇散,再置振荡器上剧烈振荡(500次/分)3分钟,于冰浴中冷却10分钟,离心(4000r/min)5分钟,取上清液9ml,置已预先装有净化材料的分散固相萃取净化管[无水硫酸镁900mg,N-丙基乙二胺(PSA)300mg,十八烷基硅烷键合硅胶300mg,硅胶300mg,石墨化炭黑90mg]中,涡旋使充分混匀,再置振荡器上剧烈振荡(500次/分)5分钟使净化完全,离心(4000r/min)5分钟,精密吸取上清液5ml,置氮吹仪上于40℃水浴浓缩至约0.4ml,加乙腈定容至1ml,涡旋混匀,用微孔滤膜(0.22μm)滤过,取续滤液,即得。
测定法 分别精密吸取供试品溶液和基质混合对照品溶液各1μl,注入气相色谱-串联质谱仪,按内标标准曲线法计算供试品中88种农药残留量。
2 液相色谱-串联质谱法
色谱条件、质谱条件 均同本法“一、定性测定方法”中液相色谱-串联质谱法项下。
对照品贮备溶液的制备、内标贮备溶液的制备、混合对照品溶液的制备、内标溶液的制备、基质混合对照品工作溶液的制备与供试品溶液的制备 均同本法“二、定量测定方法”中气相色谱-串联质谱法项下。
测定法 分别精密吸取供试品溶液和基质混合对照品溶液各1~10μl(根据检测要求与仪器灵敏度可适当调整进样量),注入高效液相色谱-串联质谱仪,按内标标准曲线法计算供试品中523种农药残留量。
【附注】(1)依据各品种项下规定的监测农药种类并参考相关农药限量规定配制对照品溶液。
(2)本法使用基质匹配标准曲线法定量,空白基质样品为经检测不含待测农药残留的同品种样品。特殊情况下,可用标准加入法对检出的农药定量。
(3)加样回收率应在70%~120%之间。在满足重复性的情况下,部分农药回收率可放宽至60%~130%。特殊情况下,可用标准加入法对回收率超出规定范围的农药定量,或在重复性满足的情况下使用回收率校正定量结果。
(4)进行样品测定时,如果检出色谱峰的保留时间与对照品一致,并且在扣除背景后的质谱图中,所选择的2对监测离子对均出现,而且所选择的监测离子对峰面积比与对照品的监测离子对峰面积比一致(相对比例>50%,允许±20%偏差;相对比例20%~50%,允许±25%偏差;相对比例10%~20%,允许±30%偏差;相对比例<10%,允许±50%偏差),则可判断样品中存在该农药。如果不能确证,选用其他监测离子对重新进样确证或选用其他检测方式的分析仪器来确证,如选用高分辨率质谱等确证手段。
(5)使用本法测定时,气相色谱-串联质谱法测定的农药,推荐选择氘代倍硫磷作为内标;液相色谱-串联质谱法测定的农药,推荐选择氘代莠去津作为内标。特殊情况下,也可选用本法推荐的其他内标。
(6)本法提供的监测离子对测定条件为推荐条件,各实验室可根据所配置仪器的具体情况作适当调整;在样品基质有测定干扰的情况下,可选用其他监测离子对测定。
(7)对于特定农药或供试品,分散固相萃取净化管中净化材料的比例可作适当调整(如对含色素较少的药材或饮片,可降低分散固相萃取净化管中石墨化炭黑的用量;测定极性较大的农药时,可降低分散固相萃取净化管中硅胶的用量),但须做加样回收试验等必要的方法学考察以确保结果准确。
(8)依据各品种项下规定的农药限量要求,在检测灵敏度满足的情况下,可对供试品溶液进行合理稀释。如省去本法氮吹浓缩步骤,取分散固相萃取净化管上清液直接测定。
(9)部分药材性质特殊,使用本法时,供试品取样量可适当调整,但一般不低于0.5g。
(10)在进行气相色谱-串联质谱法测定时,为进一步优化方法效能,供试品溶液最终定容的溶剂可由乙腈经溶剂替换为甲苯(经氮吹至近干,加入甲苯1ml溶解即可);在进行液相色谱-串联质谱法测定时,为进一步优化方法效能或部分农药色谱峰峰形,供试品溶液最终定容的溶剂可由乙腈替换为与初始流动相匹配的含水溶液。
(11)对于中成药农药残留量测定而言,可参照本法依样品的具体情况与检测要求经方法验证后取样测定。
第五法 药材及饮片(植物类)中禁用农药多残留测定法
1 气相色谱-串联质谱法
色谱条件 用(50%苯基)甲基聚硅氧烷为固定液的弹性石英毛细管柱(柱长为30m,柱内径为0.25mm,膜厚度为0.25μm)。进样口温度250℃,不分流进样。载气为高纯氦气(He)。程序升温:初始温度60℃,保持1分钟,以每分钟30℃升至170℃,再以每分钟2℃升至230℃,最后以每分钟15℃升至300℃,保持6分钟。
质谱条件 以三重四极杆串联质谱仪检测;离子源为电子轰击源(EI),离子源温度250℃。碰撞气为氮气或氩气。质谱传输接口温度250℃。质谱监测模式为多反应监测(MRM),各化合物参考保留时间、监测离子对、碰撞电压(CE)见表5。为提高检测灵敏度,可根据保留时间分段监测各农药。
2 高效液相色谱-串联质谱法
色谱条件 以十八烷基硅烷键合硅胶为填充剂(2.1mm×100mm, 1.8μm);以0.1%甲酸溶液(含5mmol/L甲酸铵)为流动相A,以甲醇-0.1%甲酸溶液(含5mmol/L甲酸铵)(95∶5)为流动相B,按表6进行梯度洗脱;流速为每分钟0.3ml,柱温为40℃。
质谱条件 以三重四极杆串联质谱仪检测;离子源为电喷雾(ESI)离子源,正、负离子扫描模式。监测模式为多反应监测(MRM),各化合物参考保留时间、监测离子对、碰撞电压(CE)见表7。为提高检测灵敏度,可根据保留时间分段监测各农药。
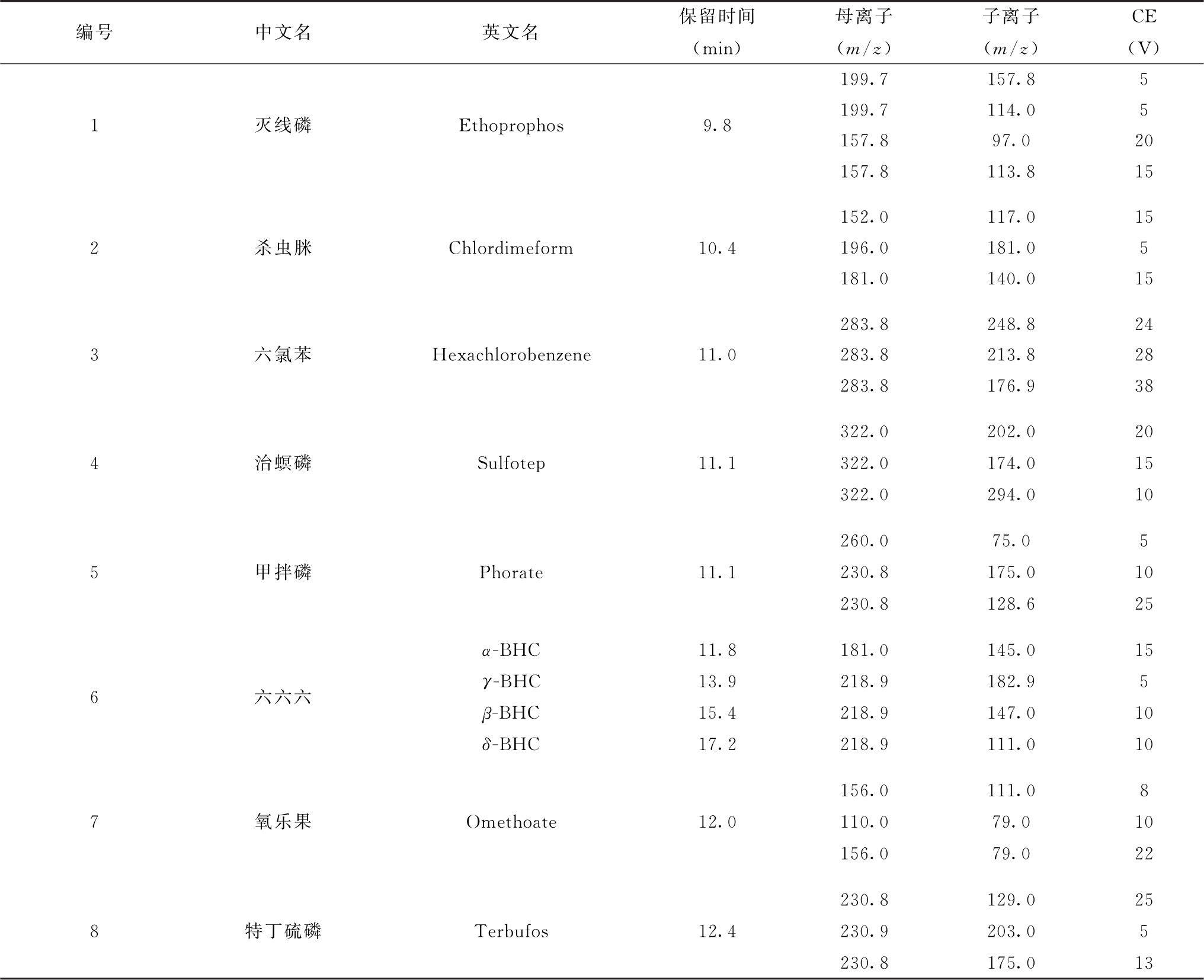
表5 各农药及相关化学品、内标化合物保留时间、监测离子对及碰撞电压(CE)参考值(GC-MS/MS部分)
续表
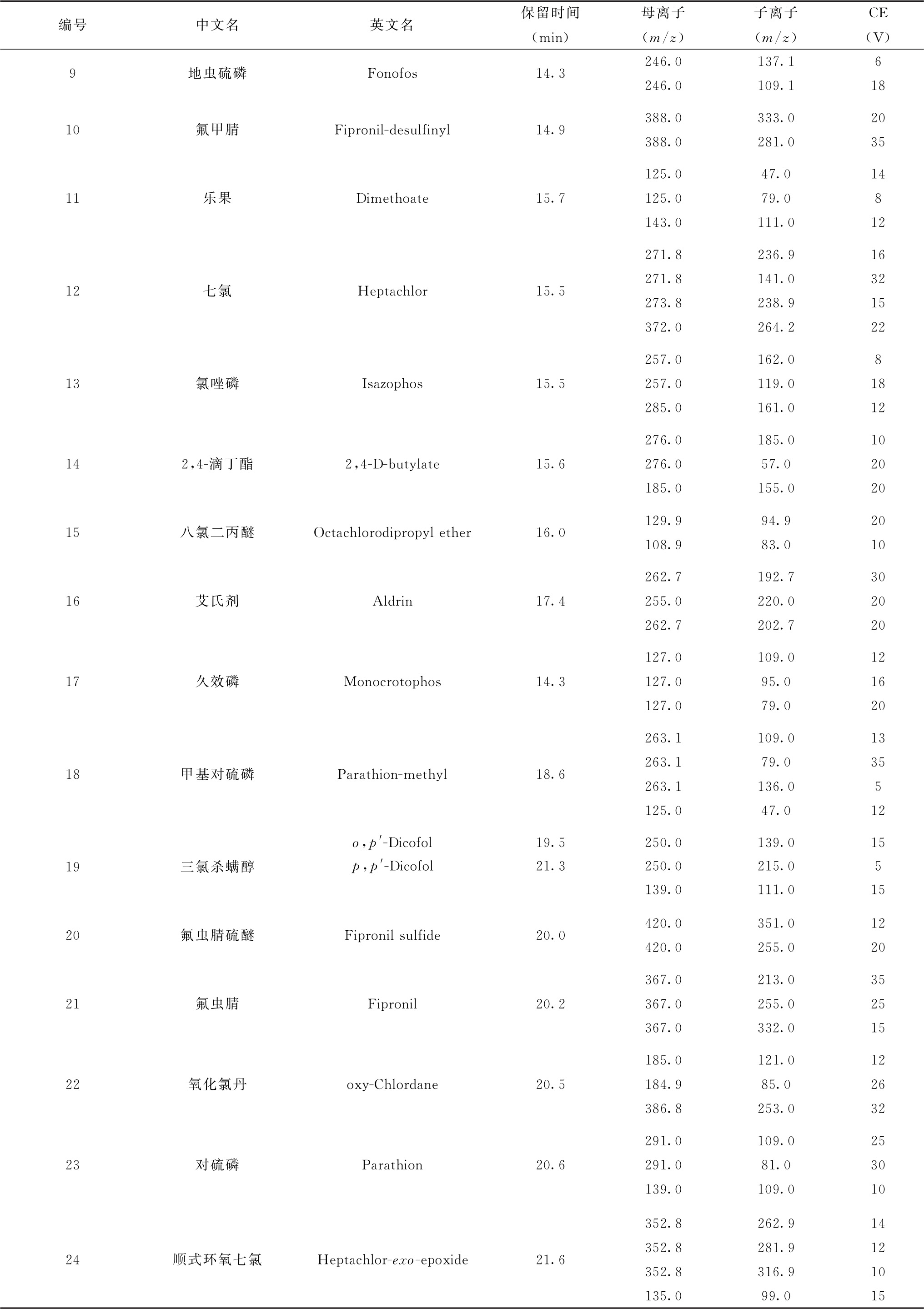
续表
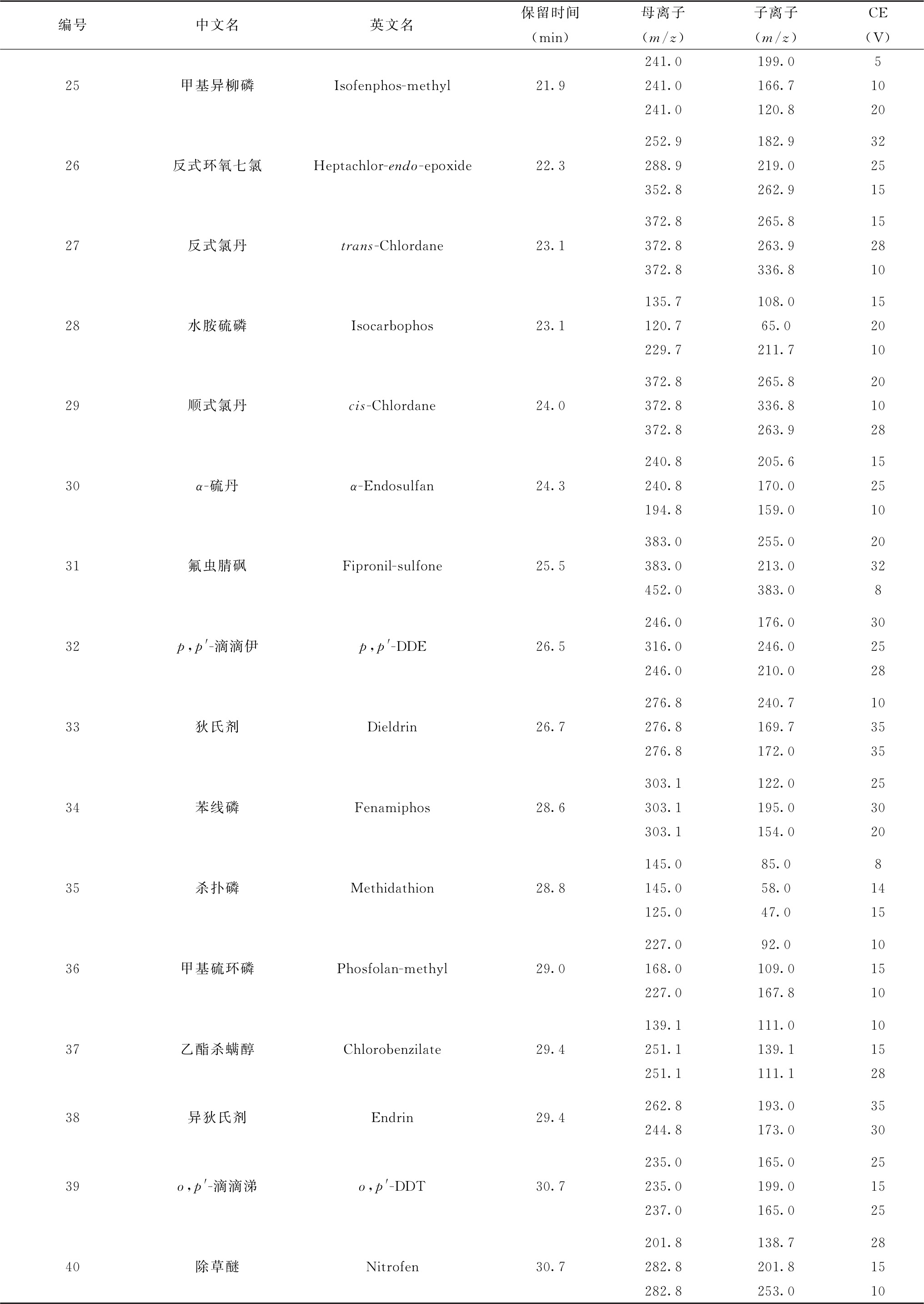
续表
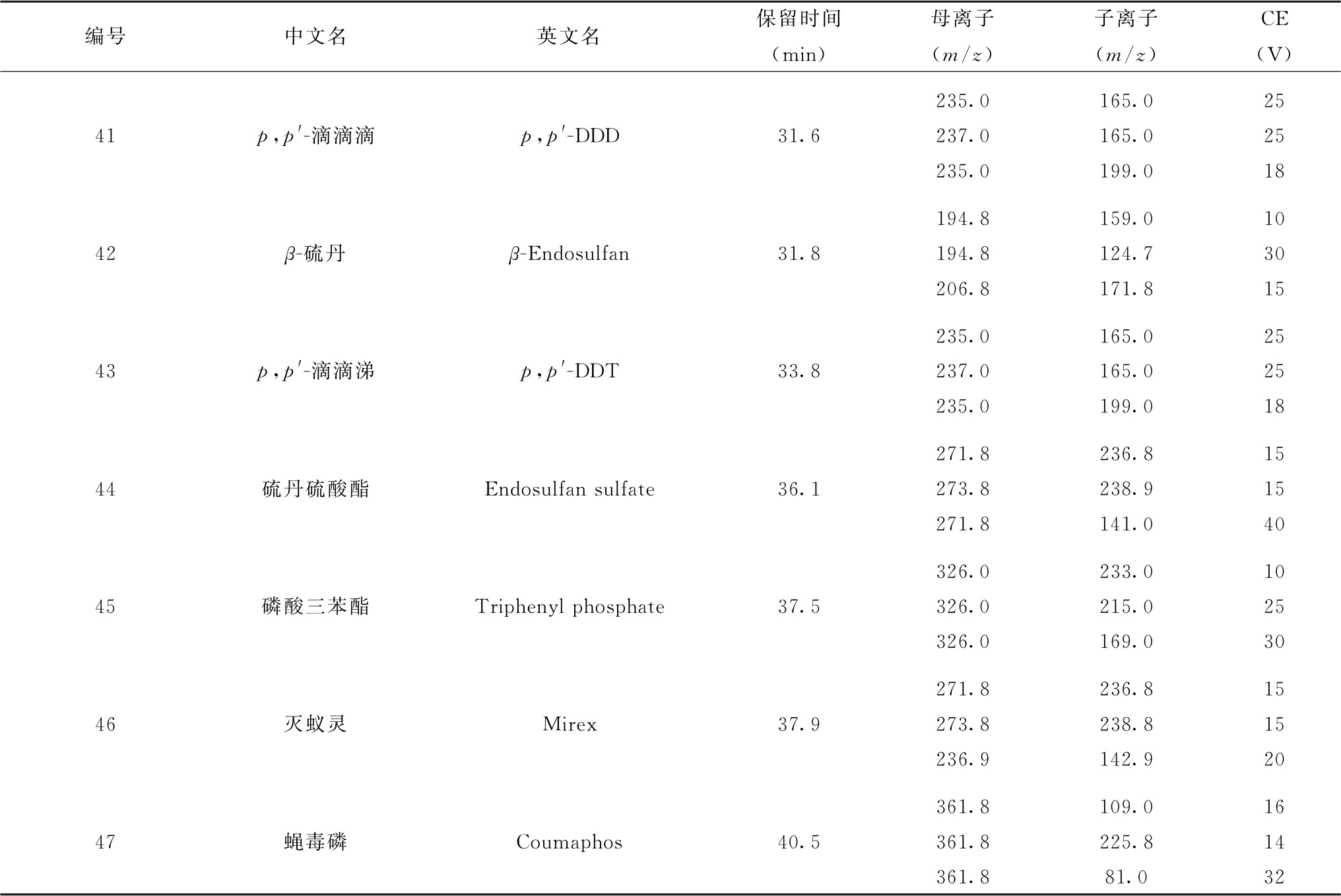
表6 流动相梯度

表7 各农药及相关化学品保留时间、监测离子对及碰撞电压(CE)参考值(LC-MS/MS部分)
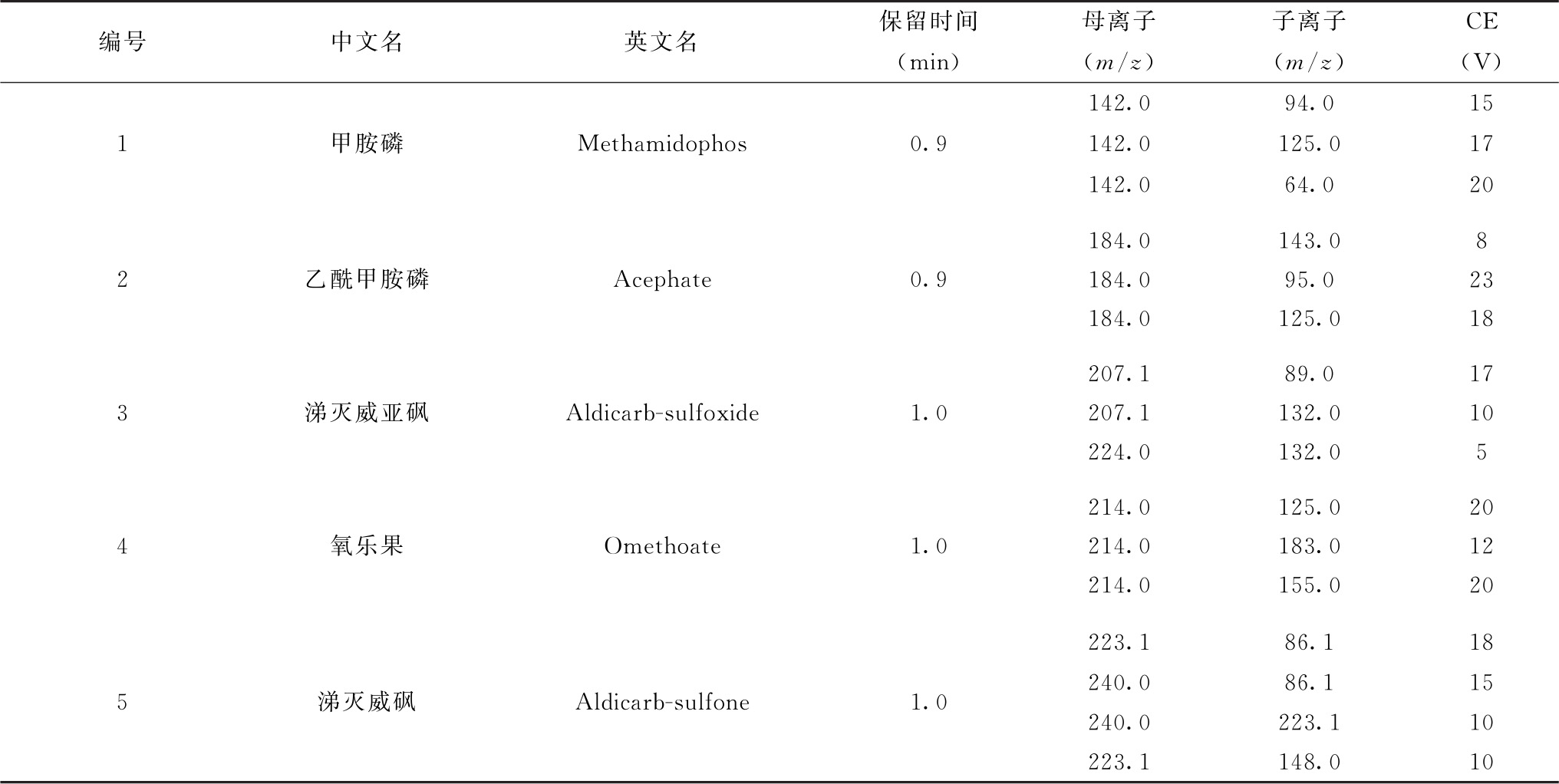
续表
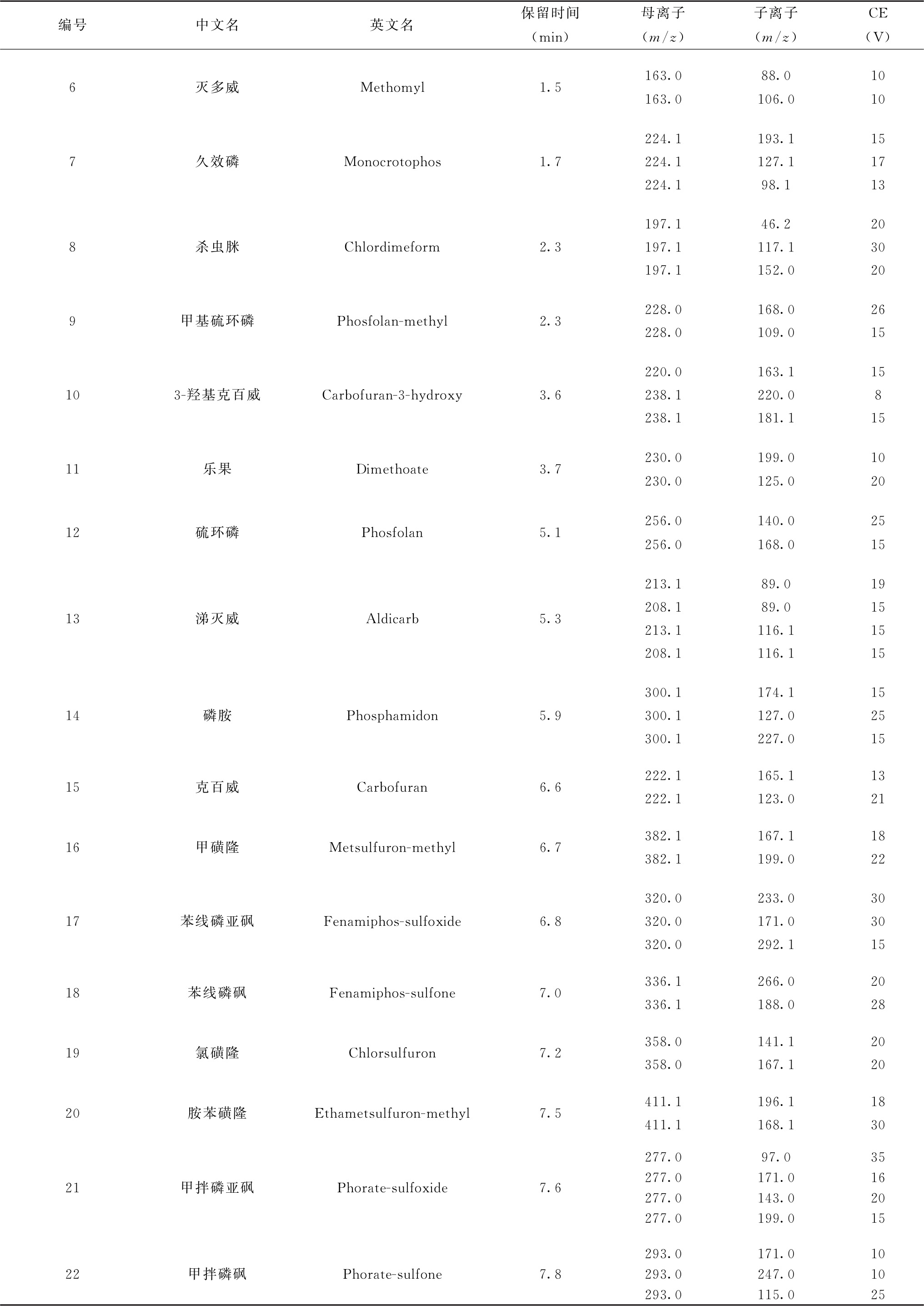
续表
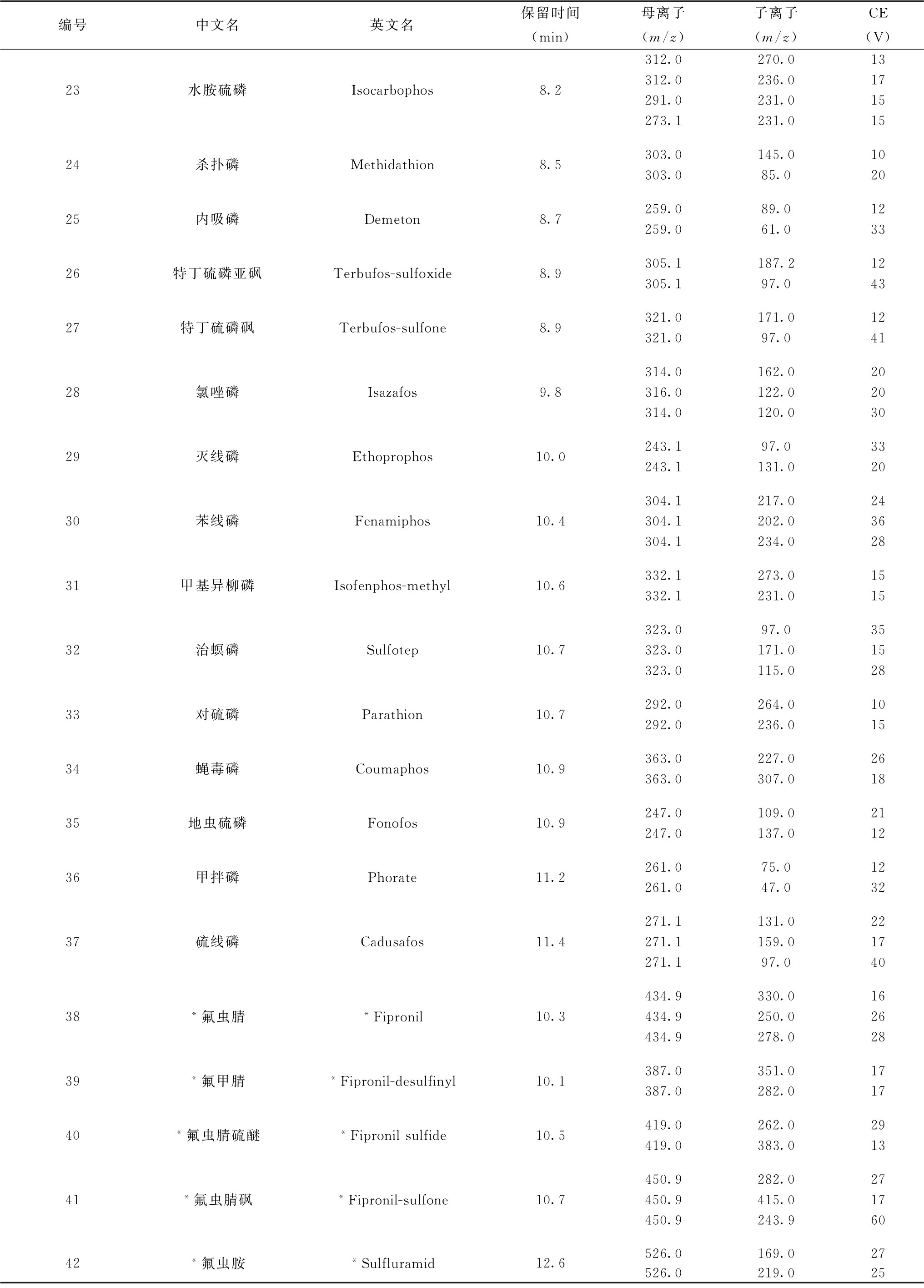
注:*表中化合物38~42使用负离子模式测定,其余化合物采用正离子模式测定。
3 对照溶液的制备
3.1 混合对照品溶液的制备 精密量取禁用农药混合对照品溶液(已标示各相关农药品种的浓度)1ml,置20ml量瓶中,用乙腈稀释至刻度,摇匀,即得。
3.2 气相色谱-串联质谱法分析用内标溶液的制备 取磷酸三苯酯对照品适量,精密称定,加乙腈溶解并制成每1ml含1.0mg的溶液,即得。精密量取适量,加乙腈制成每1ml含0.1μg的溶液。
3.3 空白基质溶液的制备 取空白基质样品,同供试品溶液相应的制备方法处理制成空白基质溶液。
3.4 基质混合对照溶液的制备 分别精密量取空白基质溶液1.0ml(6份),置氮吹仪上,40℃水浴浓缩至约0.6ml,分别精密加入混合对照品溶液5μl、10μl、20μl、50μl、100μl、200μl,加乙腈稀释至1ml,涡旋混匀,即得。
4 供试品溶液的制备
4.1 直接提取法 取供试品粉末(过三号筛)5g,精密称定,置100ml具塞离心管中,加氯化钠1g,立即摇散,再加入乙腈50ml,匀浆处理2分钟(转速不低于每分钟12 000转),离心(每分钟4000转),分取上清液,沉淀再加乙腈50ml,匀浆处理1分钟,离心,合并两次提取的上清液,减压浓缩至约5~10ml,放冷,用乙腈稀释至25ml,摇匀,即得。
4.2 快速样品处理法(QuEChERS)法 取供试品粉末(过三号筛)3g,精密称定,置50ml具塞离心管中,加入1%冰醋酸溶液15ml,涡旋使药粉充分浸润,放置30分钟,精密加入乙腈15ml,涡旋使混匀,置振荡器上剧烈振荡(每分钟500次)5分钟,加入无水硫酸镁与无水乙酸钠的混合粉末(4∶1)7.5g,立即摇散,再置振荡器上剧烈振荡(每分钟500次)3分钟,置冰浴中冷却10分钟,离心(每分钟4000转)5分钟,作为提取溶液。精密量取提取溶液9ml,置预先装有净化材料的分散固相萃取净化管(无水硫酸镁900mg,N-丙基乙二胺300mg,十八烷基硅烷键合硅胶300mg,硅胶300mg,石墨化炭黑90mg)中,涡旋使充分混匀,置振荡器上剧烈振荡(每分钟500次)5分钟使净化完全,离心(每分钟4000转)5分钟,取上清液,即得。
4.3 固相萃取法 固相萃取净化方式包括以下三种。
方式一: 精密量取直接提取法制备的供试品溶液或QuEChERS法提取溶液3~5ml,置于装有分散型净化材料的净化管(无水硫酸镁1200mg,N-丙基乙二胺300mg,十八烷基硅烷键合硅胶100mg)中,涡旋使充分混匀,再置振荡器上剧烈振荡(每分钟500次)5分钟使净化完全,离心,取上清液,即得。
方式二: 精密量取直接提取法制备的供试品溶液或QuEChERS法提取溶液3~5ml,通过亲水亲油平衡材料(HLB SPE)固相萃取柱(200mg, 6ml)净化,收集全部净化液,混匀,即得。
方式三: 精密量取直接提取法制备的供试品溶液或QuEChERS法提取溶液2ml,加在装有石墨化炭黑氨基复合固相萃取小柱(500mg/500mg, 6ml)[临用前用乙腈-甲苯混合溶液(3∶1)10ml预洗],用乙腈-甲苯混合溶液(3∶1)20ml洗脱,收集洗脱液,减压浓缩至近干,用乙腈转移并稀释至2.0ml,混匀,即得。
5 测定法
气相色谱-串联质谱法 分别精密吸取上述的基质混合对照溶液和供试品溶液各1ml,精密加入内标溶液0.1ml,混匀,滤过,取续滤液。分别精密吸取上述两种溶液各1μl,注入气相色谱-串联质谱仪,按内标标准曲线法计算,即得。
高效液相色谱-串联质谱法 分别精密吸取上述的基质混合对照溶液和供试品溶液各1ml,精密加入水0.3ml,混匀,滤过,取续滤液。分别精密吸取上述两种溶液各1~5μl,注入高效液相色谱-串联质谱仪,按外标标准曲线法计算,即得。
【附注】(1)根据待测样品基质特点和方法确认结果,选择一种最适宜的供试品溶液制备方法。对于特定农药或供试品,分散固相萃取净化管中净化材料的比例可作适当调整,但需要结合加样回收试验等必要的方法学考察以确保结果的准确性。根据规定的待测农药限量要求,在检测灵敏度满足的情况下,可以适当调整供试品溶液最终稀释倍数。供试品溶液中若检出待测农药,测定含量时,其浓度应在标准曲线的线性范围以内,且基质混合对照溶液中的基质浓度应与供试品溶液基质浓度保持一致。
(2)本法使用基质匹配标准曲线法定量,空白基质样品为经检测不含待测农药残留的同品种样品。当无法获得适宜的空白基质,或者空白基质与待测样品基质效应出现明显差异的情况下,可采用标准加入法进行定量。
(3)本法提供的监测离子对测定条件为推荐条件,各实验室可根据样品基质干扰情况和所配置仪器的具体情况作适当调整,并确定定量离子对。每个监测指标选择不少于2个监测离子对。
(4)进行样品测定时,如果检出色谱峰的保留时间与对照品一致,并且在扣除背景后的质谱图中,所选择的2个监测离子对均出现,而且所选择的监测离子对峰面积比与对照品的监测离子对峰面积比一致(相对比例>50%,允许±20%偏差;相对比例20%~50%,允许±25%偏差;相对比例10%~20%,允许±30%偏差;相对比例<10%,允许±50%偏差),则可判断样品中检出该农药。如果不能确证,选用其他监测离子对重新进样确证或选用其他检测方式的分析仪器来确证,如选用高分辨率质谱等确证手段。
(5)加样回收率应在70%~120%之间。在满足重复性要求且待测指标未检出的情况下,部分农药回收率可放宽至50%~140%。
第六法 相关药材及饮片品种中农药多残留测定法
1 气相色谱-串联质谱法
色谱条件 用(50%苯基)甲基聚硅氧烷为固定液的弹性石英毛细管柱(柱长为30m,柱内径为0.25mm,膜厚度为0.25μm)。进样口温度250℃,不分流进样。载气为高纯氦气(He)。程序升温:初始温度60℃,保持1分钟,以每分钟30℃升至170℃,再以每分钟2℃升至230℃,最后以每分钟15℃升至300℃,保持6分钟。
质谱条件 以三重四极杆串联质谱仪检测;离子源为电子轰击源(EI),离子源温度250℃。碰撞气为氮气或氩气。质谱传输接口温度250℃。质谱监测模式为多反应监测(MRM),各化合物参考保留时间、监测离子对、碰撞电压(CE)见表8。为提高检测灵敏度,可根据保留时间分段监测各农药。
表8 各农药及相关化学品保留时间、监测离子对及碰撞电压(CE)参考值(GC-MS/MS部分)
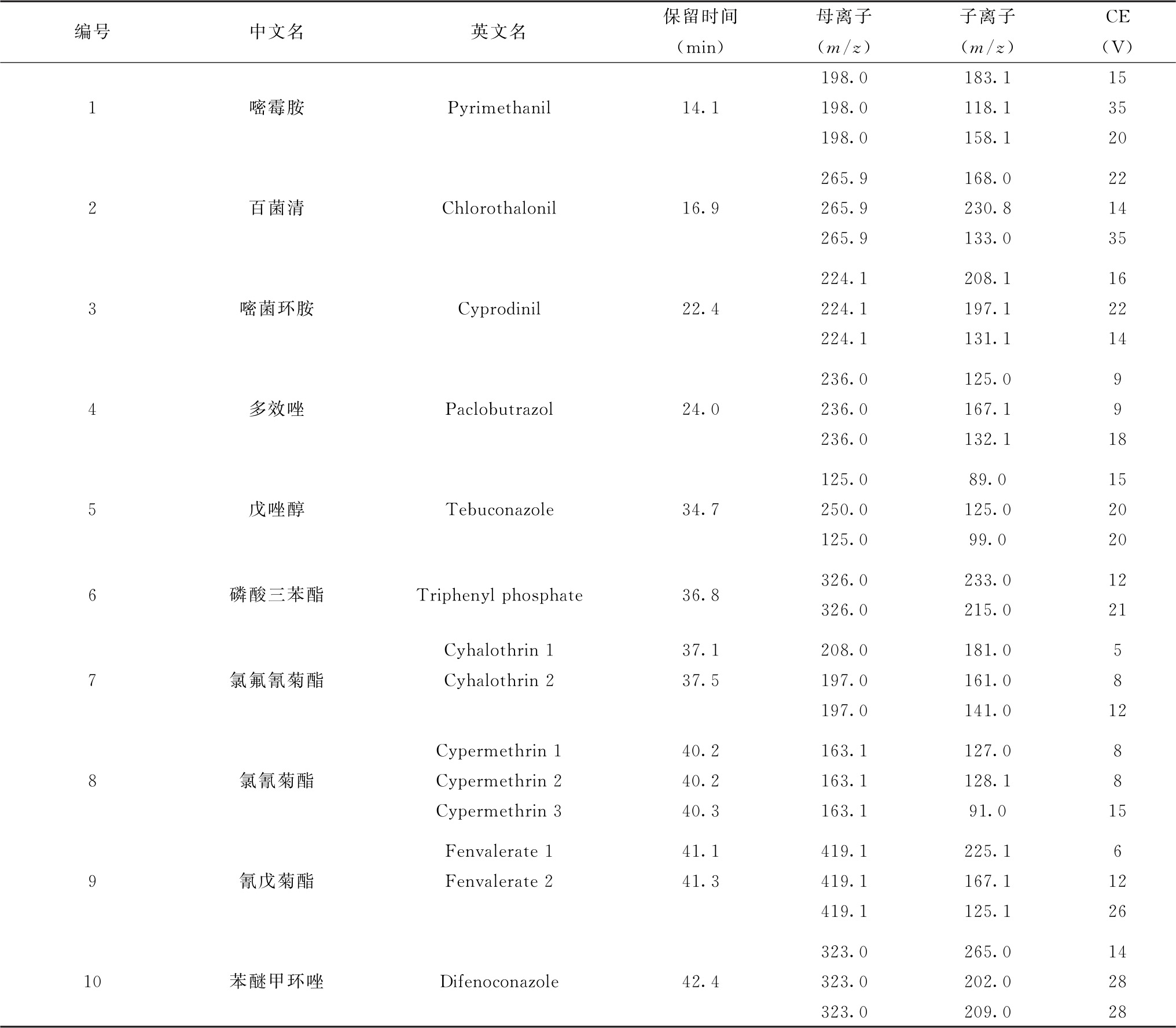
注:部分化合物含有多个异构体,表中提供了多个异构体峰的保留时间。
2 高效液相色谱-串联质谱法
色谱条件 以十八烷基硅烷键合硅胶为填充剂(2.1mm×100mm, 1.8μm);以0.1%甲酸溶液(含5mmol/L甲酸铵)为流动相A,以甲醇-0.1%甲酸溶液(含5mmol/L甲酸铵)(95∶5)为流动相B,按表9进行梯度洗脱;流速为每分钟0.3ml,柱温为40℃。
质谱条件 以三重四极杆串联质谱仪检测;离子源为电喷雾(ESI)离子源,正离子或负离子扫描模式。监测模式为多反应监测(MRM),各化合物参考保留时间、监测离子对、碰撞电压(CE)见表10。为提高检测灵敏度,可根据保留时间分段监测各农药。
表9 流动相梯度

表10 各农药及相关化学品保留时间、监测离子对及碰撞电压(CE)参考值(LC-MS/MS部分)
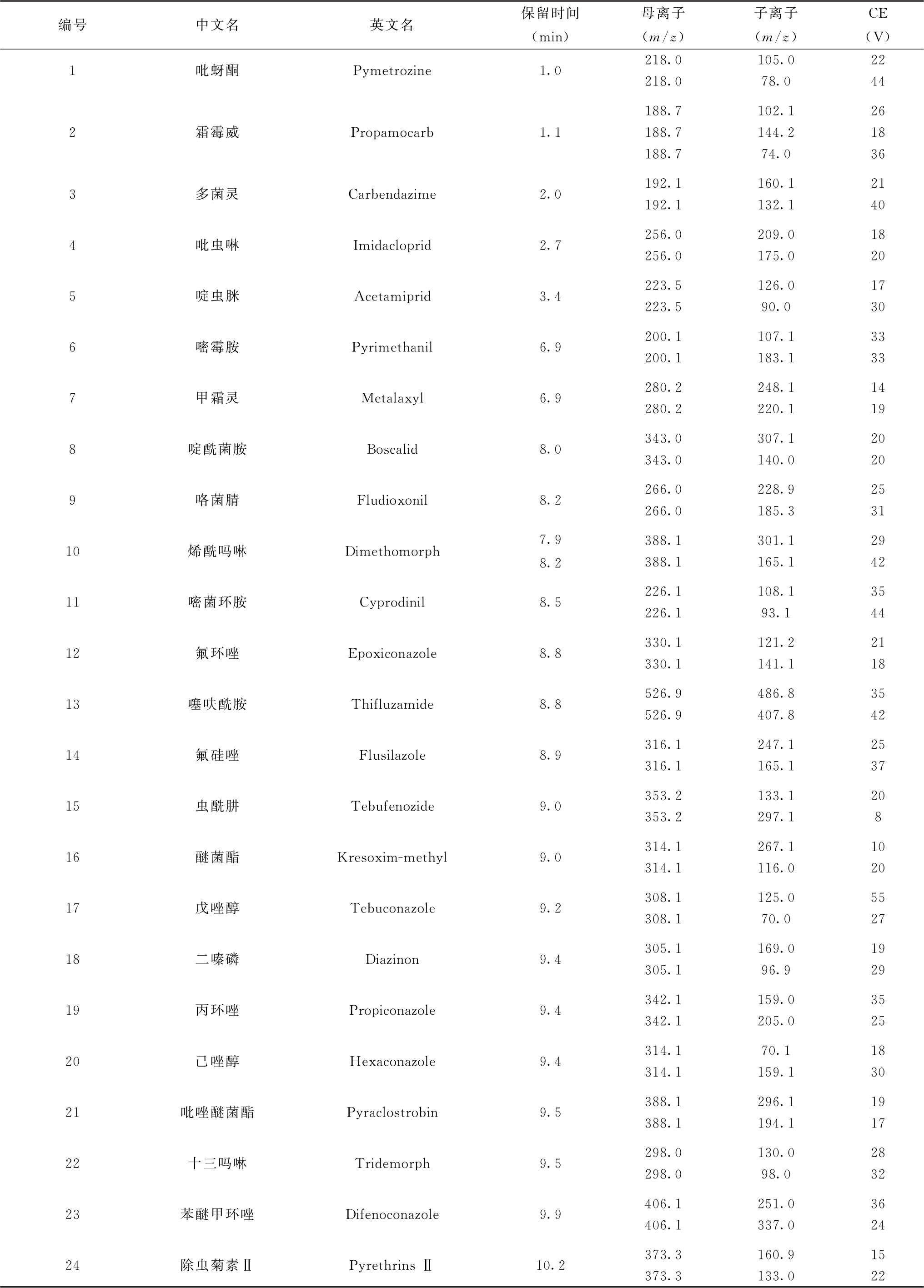
续表
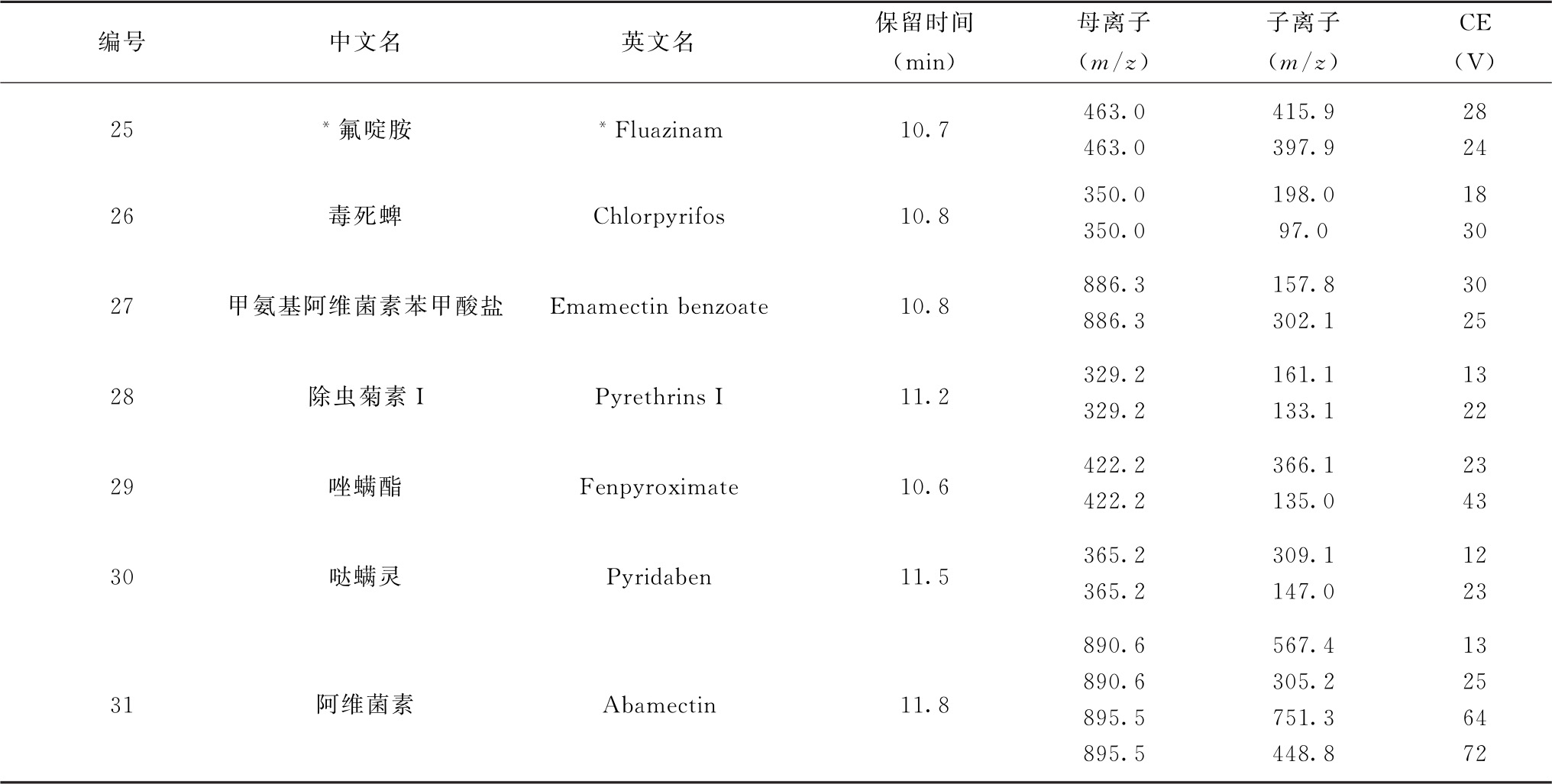
注:1.部分化合物含有多个异构体,表中提供了多个异构体峰的保留时间。
2.标记“*”的农药氟啶胺采用负离子扫描模式,其他农药均为正离子扫描模式。
3 对照溶液的制备
3.1 混合对照品溶液的制备 分别精密量取各相关农药对照品溶液适量,置20ml量瓶中,用乙腈稀释至刻度,摇匀,即得。
3.2 气相色谱-串联质谱法分析用内标溶液的制备 取磷酸三苯酯对照品适量,精密称定,加乙腈溶解并制成每1ml含1.0mg的溶液,即得。精密量取适量,加乙腈制成每1ml含0.1μg的溶液。
3.3 空白基质溶液的制备 取空白基质样品,同供试品溶液相应的制备方法处理制成空白基质溶液。
3.4 基质混合对照溶液的制备 分别精密量取空白基质溶液1.0ml(7份),置氮吹仪上,40℃水浴浓缩至约0.4ml,分别精密加入混合对照品溶液5μl、10μl、25μl、50μl、100μl、250μl、500μl,加乙腈稀释至1ml,涡旋混匀,即得。
4 供试品溶液的制备
人参、延胡索、金银花、菊花、铁皮石斛 取供试品粉末(过三号筛)5g,精密称定,置100ml具塞离心管中,加水10ml,摇匀,放置30分钟,加氯化钠5g,立刻摇散,再加入乙腈40ml,匀浆处理2分钟(转速不低于每分钟12 000转),离心(每分钟4000转)5分钟(铁皮石斛:静置),分取上清液,沉淀再加乙腈50ml,匀浆处理1分钟,离心(每分钟4000转)5分钟,合并两次提取的上清液,用乙腈稀释至100ml,摇匀,即得。
枸杞子、百合、麦冬 取供试品粉末(过三号筛,其中枸杞子需取供试品,冷冻,迅速粉碎)3g,精密称定,置50ml具塞离心管中,加入1%冰醋酸溶液15ml,涡旋使分散均匀,放置30分钟,精密加入乙腈15ml,涡旋使混匀,置振荡器上剧烈振荡(每分钟500次)5分钟,置冰浴中放置20分钟,加入无水硫酸镁与无水乙酸钠的混合粉末(4∶1)7.5g,立即摇散,再置振荡器上剧烈振荡3分钟,离心(每分钟4000转)5分钟,精密吸取上清液5ml,用乙腈稀释至20ml(百合、麦冬用乙腈稀释至10ml),摇匀,即得。
三七、浙贝母、川贝母、湖北贝母、伊贝母、平贝母、白术 取供试品粉末(过三号筛)3g,精密称定,置50ml具塞离心管中,加入1%冰醋酸溶液15ml,涡旋使分散均匀,放置30分钟,精密加入乙腈15ml,涡旋使混匀,置振荡器上剧烈振荡(每分钟500次)5分钟,置冰浴中放置20分钟,加入无水硫酸镁与无水乙酸钠的混合粉末(4∶1)7.5g,立即摇散,再置振荡器上剧烈振荡3分钟,离心(每分钟4000转)5分钟。精密量取上清液9ml,置预先装有净化材料的分散固相萃取净化管(无水硫酸镁900mg,N-丙基乙二胺300mg,十八烷基硅烷键合硅胶300mg,硅胶100mg,石墨化炭黑45mg)中,涡旋使充分混匀,置振荡器上剧烈振荡(每分钟500次)5分钟使净化完全,离心(每分钟4000转)5分钟,精密吸取上清液5ml,用乙腈稀释至20ml,摇匀,即得。
5 测定法
气相色谱-串联质谱法 分别精密吸取上述的基质混合对照溶液和供试品溶液各1ml,精密加入内标溶液0.1ml,混匀,滤过,取续滤液。分别精密吸取上述两种溶液各1μl,注入气相色谱-串联质谱仪,按内标标准曲线法计算,即得。
高效液相色谱-串联质谱法 分别精密吸取上述基质混合对照溶液和供试品溶液各1ml,精密加水0.3ml,混匀,滤过,取续滤液。分别精密吸取上述两种溶液各1~5μl,注入高效液相色谱-串联质谱仪,按外标标准曲线法计算,即得。
【附注】(1)根据规定的待测农药限量要求,在检测灵敏度满足的情况下,可以适当调整供试品溶液最终稀释倍数。供试品溶液中若检出待测农药,测定含量时,其浓度应在标准曲线的线性范围以内。
(2)本法使用基质匹配标准曲线法定量,空白基质样品为经检测不含待测农药残留的同品种样品。当无法获得适宜的空白基质,或者空白基质与待测样品基质效应出现明显差异的情况下,可采用标准加入法进行定量。
(3)本法提供的监测离子对测定条件为推荐条件,各实验室可根据样品基质干扰情况和所配置仪器的具体情况作适当调整,并确定定量离子对。每个监测化合物选择不少于2个监测离子对。
(4)进行样品测定时,如果检出色谱峰的保留时间与对照品一致,并且在扣除背景后的质谱图中,所选择的2个监测离子对均出现,而且所选择的监测离子对峰面积比与对照品的监测离子对峰面积比一致(相对比例>50%,允许±20%偏差;相对比例20%~50%,允许±25%偏差;相对比例10%~20%,允许±30%偏差;相对比例<10%,允许±50%偏差),则可判断样品中检出该农药。如果不能确证,选用其他监测离子对重新进样确证或选用其他检测方式的分析仪器来确证,如选用高分辨率质谱等确证手段。
(5)加样回收率应在70%~120%之间。在满足重复性要求且待测农药未检出的情况下,部分农药回收率可放宽至50%~140%。
第七法 药材及饮片中二硫代氨基甲酸盐类农药残留量测定法
色谱、质谱条件 以6%氰丙基苯基-94%二甲基聚硅氧烷为固定液的弹性石英毛细管柱(柱长为30m,柱内径为0.25mm,膜厚度为1.4μm),载气为高纯氦气,柱流速1.5ml/min;进样口温度为180℃,分流进样,分流比5∶1。程序升温:初始温度40℃,保持6分钟,以30℃/min升至250℃,保持2分钟。
以三重四极杆串联质谱仪检测;离子源为电子轰击源(EI),离子源温度250℃。碰撞气为氮气或氩气。质谱传输接口温度250℃。监测模式为多反应监测(MRM),监测离子对(碰撞电压CE)为76.0→32.0(27V),76.0→44.0(32V)。
系列对照品溶液的制备 取二硫化碳对照品,精密称定,用异辛烷分别稀释制成每1ml含0.05μg、0.1μg、0.2μg、0.5μg、1.0μg、2.0μg的溶液,作为系列浓度对照品溶液(临用新制)。
供试品溶液的制备 取供试品粉末(过三号筛)1g,精密称定,置顶空瓶中,精密加入异辛烷3ml,加盐酸-氯化亚锡溶液(取二水合氯化亚锡7.5g,加盐酸215ml使溶解,加水至500ml,摇匀)5ml,立即密封,振摇,使分散。置80℃水浴中1小时,时时振摇。取出,冷却,摇匀,离心(每分钟5000转)3分钟,取上层有机相,即得。
测定法 分别精密吸取上述系列对照品溶液1μl,注入气相色谱-串联质谱仪,测定,以峰面积为纵坐标,进样浓度为横坐标,绘制标准曲线。另精密吸取供试品溶液1μl,注入气相色谱-串联质谱仪,测定,按外标标准曲线法计算二硫化碳(CS2)的含量,即得。