0862 元素杂质
存在于药品中的元素杂质有多种来源,生产过程中使用的原料药、辅料、生产设备、水和包装材料等均可能引入元素杂质,贮存过程中包装材料中的元素杂质还可能发生迁移而被引入药品中。这些元素杂质可能是有意添加引入(如原料药或辅料合成过程中有意添加的催化剂残留),也可能是无意引入(如与生产设备或包装系统相互作用产生的杂质或药品各个组分中存在的杂质)。为了治疗作用而有意添加到药品中的元素不属于元素杂质。
因元素杂质不能为患者提供任何治疗作用,某些元素杂质甚至有一定毒性,所以它们在药品中的量需被控制在可接受的限度范围内。本通则提供评估和控制药品中元素杂质的有关依据和确认元素杂质种类及其限度的方法,为元素杂质测定方法的选择、建立、验证和使用提供指导。
本通则不适用于中药、放射性药物、疫苗、细胞代谢产物、DNA产品、过敏原提取物、细胞、全血、血细胞成分或包括血浆及血浆衍生产品在内的血液衍生产品和非体循环透析液;也不适用于基于基因(基因治疗)、细胞(细胞治疗)和组织(组织工程)的药品。
本通则规定的限度不直接适用于原料药和辅料。但为使药品中的元素杂质能符合规定,制剂生产企业可以使用原料药或辅料生产企业提供的元素杂质测定数据或者风险评估报告,用于证明最终制剂符合本通则的限度要求。原料药和辅料生产企业选择进行风险评估的元素,可依照表1进行。对某些天然来源的原料药和辅料,因其含有自然界与生俱来的元素,必须在风险评估中加以考虑。
1 风险评估中建议考察的元素
考虑元素杂质毒性和出现在药品中的相对可能性,其可以分为三类。根据药品中元素杂质分类、是否是有意添加引入和给药途径,风险评估中建议考察的元素杂质见表1。
2 形态
药品中元素形态是元素在药品中存在的化学形式(包括同位素组成、电子态或氧化态、配合物或分子结构)。元素的不同形态可能具有不同的毒性,应予以关注。当某种元素不同形态的毒性已知时,采用预期出现在药品中的形态的毒性信息来确定PDE。风险评估时如果某种元素杂质是用药品中该元素总量来评估是否符合PDE,一般情况下不要求提供该元素的形态信息,但当发现该元素实际的形态比用于确定表2中PDE的形态(见ICH Q3D附录3)具有更高或更低毒性时,可提供该元素的形态信息以证明使用更低或更高水平PDE的合理性。
3 允许暴露量
口服、注射、吸入和皮肤给药四种给药途径的每日允许暴露量(PDE)见表2。在本通则中,PDE以μg/d为单位,表示药品中某种元素的最大日允许摄入量。
日最大剂量不超过2L的注射剂,可使用日最大剂量由PDE计算允许浓度。说明书规定或临床实践确定日剂量超过2L的药品(如0.9%氯化钠溶液、葡萄糖注射液、全胃肠外营养液、灌洗液等),可使用2L体积由PDE计算允许浓度。
表1 风险评估中建议考察的元素
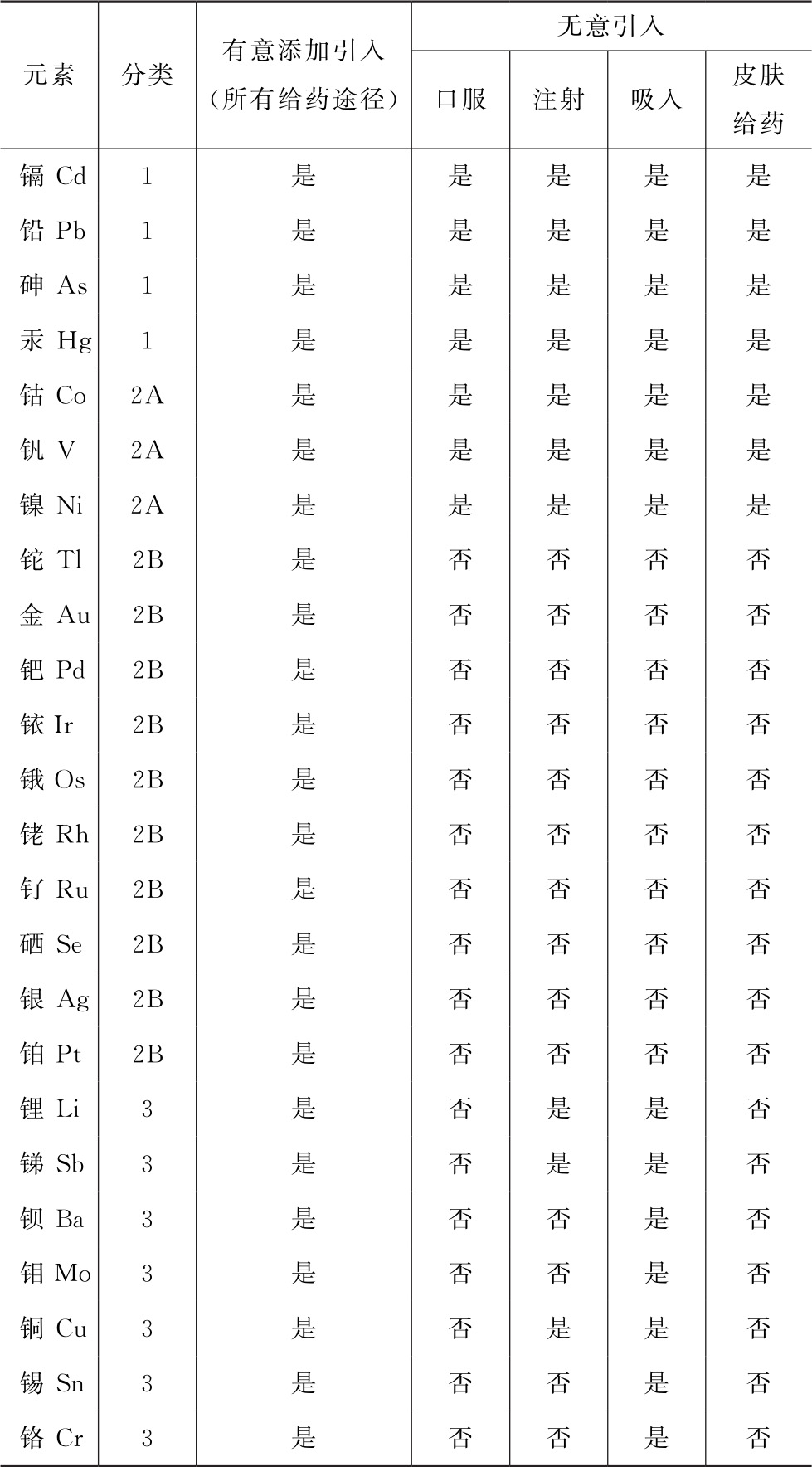
注:①1类元素是对人体有害元素,在药品生产中禁用或限制使用。
②2类元素通常被认为是给药途径依赖型的人体有害元素。根据它们出现于药品中的相对可能性,进一步分成2A和2B亚类。
③3类元素口服给药途径的毒性相对较低(PDE高,通常>500μg/d),风险评估中一般不需考虑,但在吸入和注射给药途径时大部分元素PDE小于500μg/d,风险评估中仍需考虑。
④此表来源于ICH Q3D。
皮肤给药途径的PDE适用于治疗皮肤擦伤或者其他快速愈合急性损伤的药品,但不适用于治疗表皮的基底细胞层受到实质性破坏的皮肤给药药品。对于需要使药品与真皮接触的适应症(例如,皮肤溃疡、Ⅱ度以上的烧伤、大疱疮、大疱性表皮松解症等),通常可以在注射给药途径PDE基础上,根据该药品的具体情况进行修正,论证合理的PDE。此外,皮肤给药途径的药品还需考虑元素杂质的致敏性。对于致敏元素Ni和Co,应同时满足PDE及皮肤和透皮给药途径浓度限度(CTCL),以减少过敏个体引起皮肤反应的可能性。Ni和Co的CTCL均为35μg/g。对于其他元素杂质,引起过敏反应的阈值大约等于皮肤给药途径PDE或者远大于皮肤给药途径PDE时,不需要额外控制。
表2 元素杂质的每日允许暴露量(PDE) 单位:μg/d
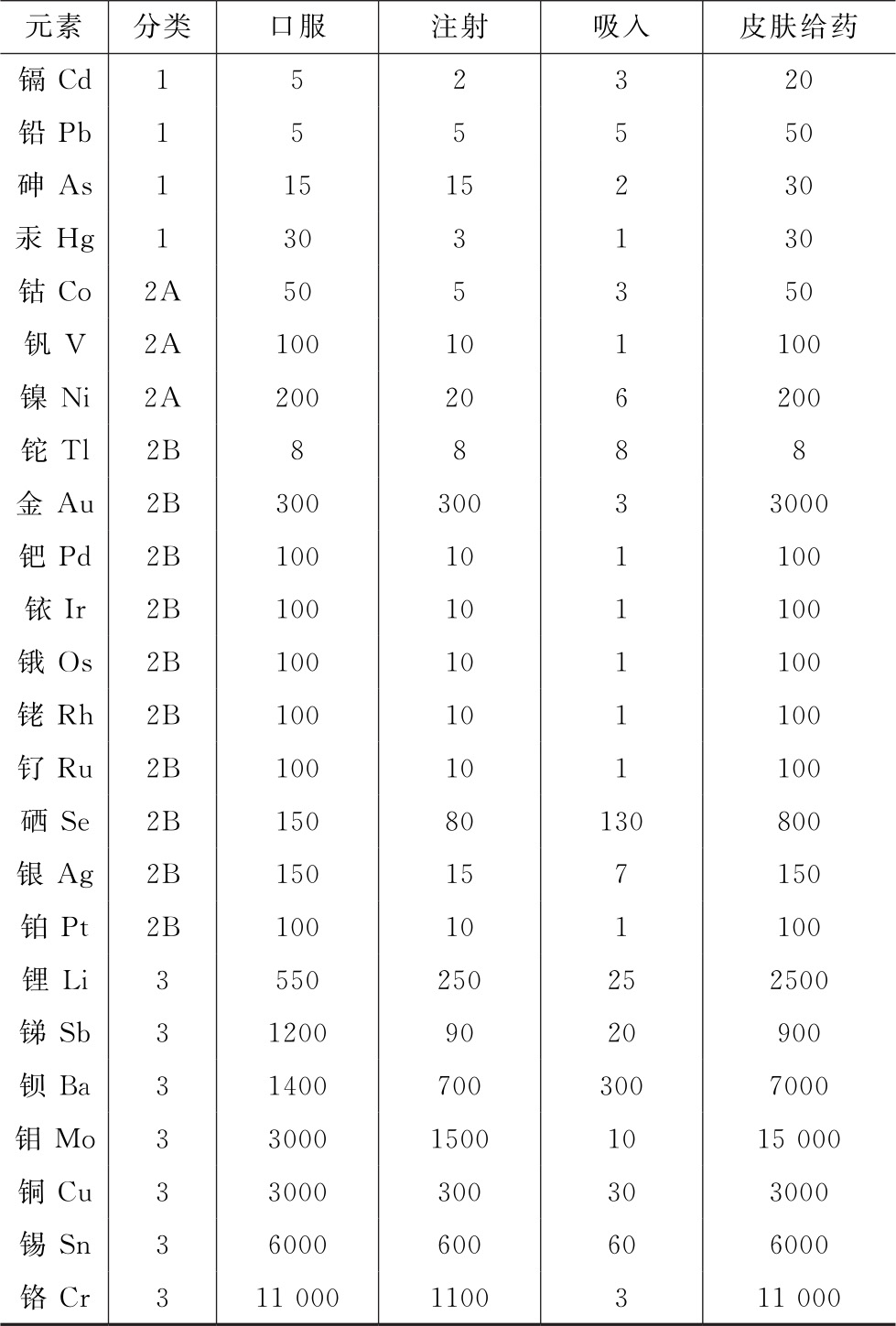
注:此表来源于ICH Q3D,各元素PDE以ICH Q3D最新版为准。
4 限度确认方法
根据不同需求可任意选择以下3种方法进行限度确认。当选择下述方法2或方法3进行限度确认时,均需考虑由包装系统和生产设备引入药品中的元素杂质,如果在风险评估过程中,已确定包装系统和生产设备对药品的元素杂质水平没有影响,则无需考虑;当包装系统和生产设备对药品的元素杂质水平有影响时,需考虑以估计日摄入量的形式从PDE中扣除这些来源的影响后再采用方法2或方法3进行限度确认。
方法1制剂分析法 测定制剂中每种元素的浓度,根据每日最大剂量计算每种元素杂质的每日摄入总量,再与各元素的PDE比较。
PDE≥测得元素浓度(μg/剂量单位)×每日最大剂量(剂量单位/天)
注:剂量单位指药品每日摄入量的单位,如g、ml等。
除另有规定外,每种元素杂质的每日摄入总量应不得高于该元素的PDE。
方法2组分加和法 分别加和制剂中所有组分(原料药及辅料)所含元素杂质的量(单位μg/d):
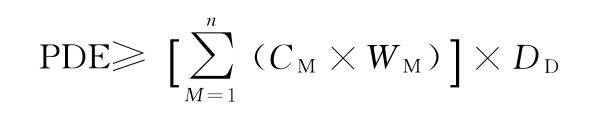
式中 M为制剂中的某一组分(原料药或辅料);
CM为组分中的元素浓度,μg/g;
WM为每剂量单位中组分的质量,g/剂量单位;
DD为每日最大剂量单位数,剂量单位/天。
除另有规定外,每种元素杂质加和结果应不得高于PDE。
方法3单组分限度法 原料药和辅料中元素杂质的可接受水平取决于其最终用途。表3中提供的数据是以10g/d作为最大日剂量计算的原料药和辅料的通用限度。对于日剂量不超过10g的药品,如果处方中所有原辅料中的元素杂质含量均不超过表3中所示的限度,则这些组分可以任意比例使用,无需进一步计算。这些数据用作通用限度,为制剂企业和原辅料供应商提供参考和帮助。
表3 日剂量不超过10g的药品中单个组分通用的元素杂质限度 单位:μg/g
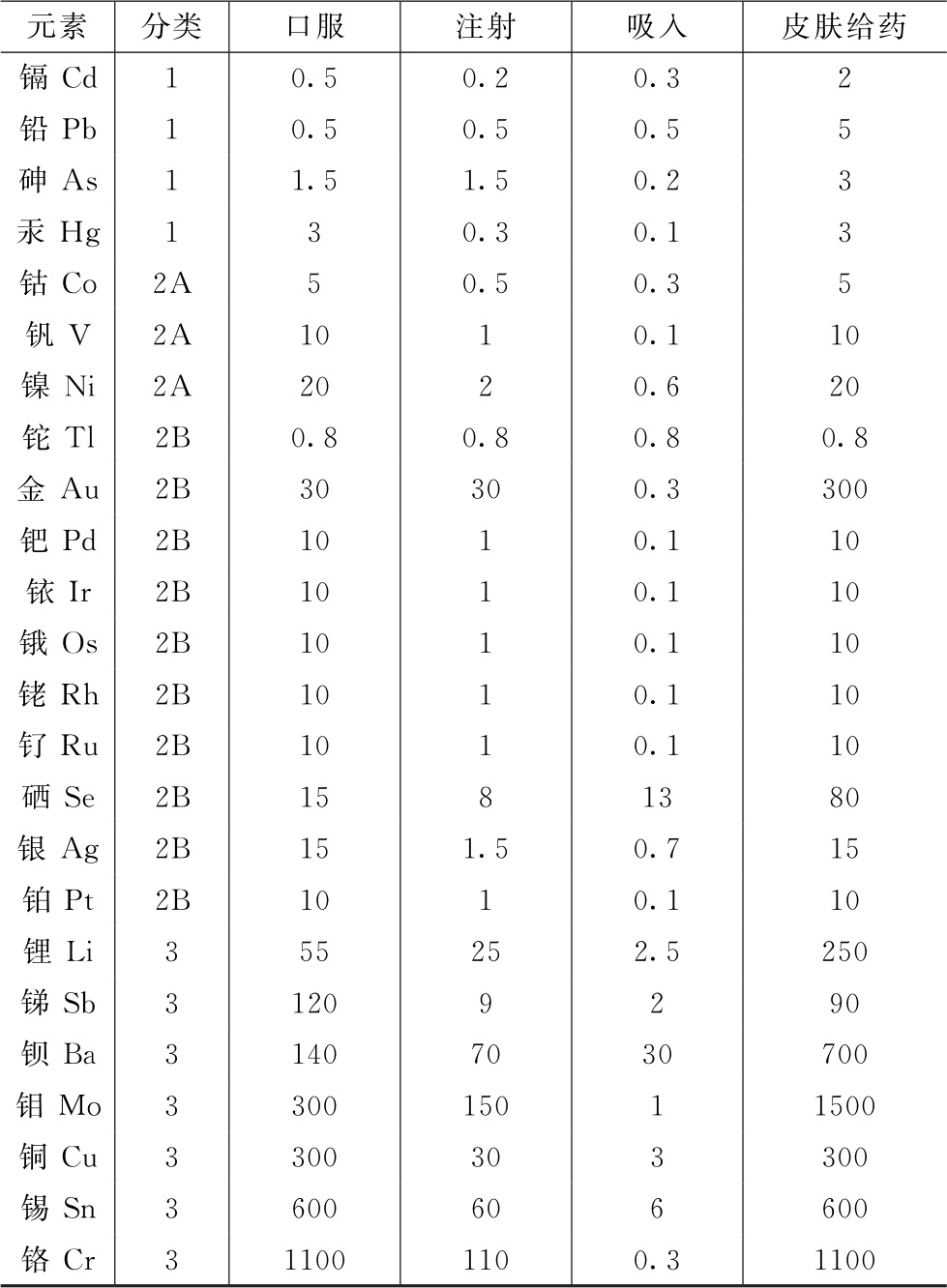
注:此表来源于ICH Q3D,各元素通用限度以ICH Q3D最新版为准。
当制剂的每日最大摄入量明确时,制剂中各组分可以按方法1项下公式计算并确认限度,如果各组分每种元素杂质的每日摄入总量均不高于该元素的限度,则所有这些组分都可以在该药品中以任意比例使用。
5 评估
潜在元素杂质的风险评估存在以下两种可能结果:
(1)风险评估过程未发现任何潜在的元素杂质。应记录风险评估结论和支持性信息及数据。
(2)风险评估过程发现一个或多个潜在的元素杂质。对于该过程中发现的任何元素杂质,风险评估均需考察元素杂质的来源多样性,并记录评估结论和支持性信息。
原料药、辅料、包装材料和生产设备供应商提供的关于潜在元素杂质的信息有助于药品生产企业开展元素杂质风险评估。支持该风险评估的数据来源包括但不限于:先验知识、公开发表的文献、相似工艺的数据、供应商信息或数据、制剂组分的检验、制剂的检验等。
影响药品中潜在元素杂质水平的因素也需在风险评估中予以考虑。这些因素包括但不限于:在后续工艺过程中清除元素杂质的有效性、元素的天然丰度(对于无意引入的元素尤为重要)、对于特定来源的元素杂质浓度范围的先验知识、制剂的组成等。
6 控制阈值
元素杂质控制应考察检测到的元素杂质水平相对于其PDE的显著性。将药品中元素杂质PDE(以及Ni和Co的CTCL)的30%定义为控制阈值,作为元素杂质水平显著性的衡量指标。控制阈值可用于判断药品中的元素杂质是否需要额外的控制。
如果药品中某个元素杂质水平一直低于控制阈值,只要对数据进行适当的评估并表明已对元素杂质进行足够的控制,则不再需要额外的控制。
如果风险评估无法表明某个元素杂质水平一直低于控制阈值,就需要建立控制方法以保证药品中元素杂质水平不超过PDE。
7 元素杂质的控制
元素杂质的控制是药品整体控制策略的一部分,用以确保元素杂质不超过PDE。当元素杂质水平超过控制阈值时,需采取额外的手段来确保元素杂质水平不超过PDE。控制药品中元素杂质能够采用的方法包括但不限于:
(1)调整相关生产工艺,通过特定或非特定的纯化步骤将元素杂质降低至控制阈值以下;
(2)实施工艺过程的中游或上游控制,将药品中元素杂质的浓度限制在控制阈值以下;
(3)建立辅料或物料(如合成中间体)的元素杂质标准限度;
(4)建立原料药的元素杂质标准限度;
(5)建立制剂的元素杂质标准限度;
(6)选择合适的包装材料;
(7)对药品中元素杂质进行定期检测。
有关元素杂质控制的证明性材料包括但不限于:风险评估总结、能支持结论的数据和确定元素杂质限度控制的具体方法。
8 测定方法
任何可以满足质量控制要求的方法均可用于元素杂质的测定。检测方法包括但不限于电感耦合等离子体质谱法(通则0412)、电感耦合等离子体原子发射光谱法(通则0411)、原子吸收分光光度法(通则0406)、X射线荧光光谱法(通则0461)、重金属检查法(通则0821)、硒检查法(通则0804)、砷盐检查法(通则0822)等。上述方法通常测定的是不同形态元素的总量,若需区分元素的形态,可采用电感耦合等离子体质谱法与分离技术如液相色谱法联用的方法,或其他适宜的方法进行测定。
各测定方法的具体操作可参考本版药典通用技术要求及相关的指南和标准操作规范。建立的任何一种测定方法,均需经过分析方法验证,以证明分析方法满足预期的质量控制目的。
8.1 供试品制备
液体供试品根据其基质、有机物含量和待测元素含量等情况,可选用直接测定、经稀释或浓缩后测定、消解后测定等不同方式。固体供试品一般需先制备成供试品溶液后方可用于测定,制备方法包括采用如水性溶液、有机溶剂或混合溶液进行溶解、稀释等。某些供试品不能直接制成溶液的,或制成溶液后存在基质干扰等因素影响测定的,或基于测定方法要求需转换形态的,常需经消解后再制成适当的溶液。选择消解方法时,应考虑供试品的性质、待测元素的性质及其含量等因素。消解方法包括密闭容器消解法(常用微波消解法)和敞口容器消解法(包括电热板湿法消解、干法灰化等),其中微波消解法为最常用的消解方法之一,敞口容器消解法不适合挥发性元素的测定。
采用电感耦合等离子体质谱法、电感耦合等离子体原子发射光谱法或原子吸收分光光度法进行测定时,制备方法的选择可参考图1。实验用溶剂或稀释剂优先使用水性溶液,最常用的为稀酸溶液,如制成一定浓度的硝酸、盐酸、氢氟酸、高氯酸、硫酸水溶液等。硝酸带来的干扰最小,是首选酸。当使用氢氟酸时须先与仪器厂家确认所用仪器对氢氟酸的耐受性。此外,还可使用稀释的过氧化氢溶液、稀碱溶液、混合的酸或碱、有机溶剂(浓的、稀释的或混合的)等。由于实验过程中使用的任何试剂都可能引入基质效应或产生干扰,推荐使用高纯度的酸、碱、过氧化氢或有机试剂,实验用水必须为去离子水。一般情况下,供试品溶液需是澄清的,若特殊情况需要浊液进样,应经过充分验证。供试品制备时应同时制备空白试剂样品。
采用原子吸收分光光度法进行测定时,基于增强待测易挥发元素的稳定性、提高基体的挥发性、改变化学组成以提高分析物的原子化效率等因素考虑,可在供试品溶液中加入适当浓度的基体改进剂,但需注意基体改进剂可能由于试剂不纯等因素带来新的干扰。常用的基体改进剂有磷酸二氢铵、磷酸氢二铵、硝酸钯、硝酸镁、硝酸铵等。
采用X射线荧光光谱法测定时,液体供试品和固体供试品大多数情况下均可直接取样测定,但需注意供试品的均匀性。对于有包衣的制剂等均匀性不佳的供试品,建议粉碎并混合均匀后进行测定,有时可能还需要进一步压片后再进行测定。
取样应均匀并有代表性。供试品制备时应避免污染,包括制备过程(粉碎、分样、溶解、稀释、浓缩、消解等)、实验室环境、试剂(水)质量、器皿等。痕量分析时,实验用器皿可能需要经过一定的预处理以防止离子污染,可采用一定浓度的硝酸或硝酸-盐酸混合液浸泡后用去离子水反复冲洗干净,或用其他方法去除器皿中易溶出的离子。此外,必须注意防止元素杂质在容器表面吸附。
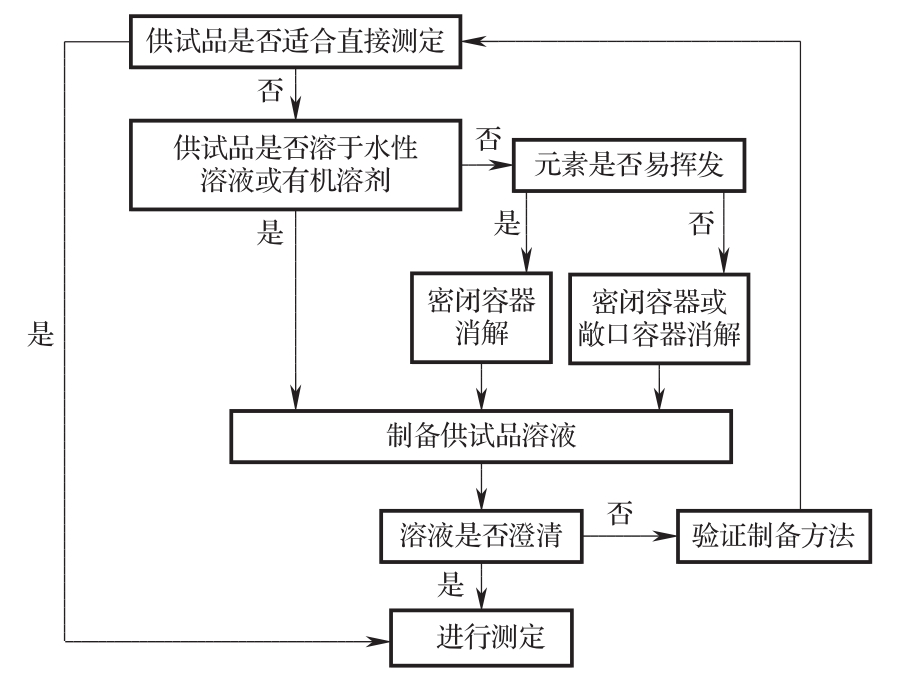
图1 供试品制备参考决策图
8.2 方法选择
如适用,可选择任一满足预期质量控制目的的方法测定元素杂质,其中电感耦合等离子体质谱法和电感耦合等离子体原子发射光谱法已成为同时测定多种元素杂质、进行元素杂质风险评估的常用方法。由于在灵敏度、专属性、线性范围、多元素同时测定等方面的优势,电感耦合等离子体质谱法通常还是同时测定多种痕量元素杂质的首选方法。
元素杂质控制方法的选择主要取决于对元素杂质的控制要求、待测元素的性质和供试品的基质,可采用定量或限度检查等方法控制药品中的杂质量。定量方法包括外标法、内标法、标准加入法和标准曲线法等。根据方法精密度、准确度、基质干扰等情况,通常采用标准曲线法,也可采用外标法、内标法或标准加入法进行定量。如采用外标法,当系统适用性、准确度或精密度等达不到要求时,可考虑选择内标法或标准加入法,以减小供试品制备和测定时因干扰或损失带来的影响。计算供试品待测元素含量时必须采用适宜方法扣除试剂空白值。
仪器分析参数的选择和设置可参考仪器制造商的说明书或操作指南。必须注意背景干扰、基质效应、记忆效应和其他原因对测定的干扰。
8.3 方法验证
建立的分析方法必须通过方法验证来证明满足其预期的目的。方法验证应遵循分析方法验证指导原则(指导原则9101),准确度和精密度试验要求以本通则为准。方法验证时,用于评价准确度和精密度的试验样品应采用与待测供试品相同的方式制备、测定。
(1)专属性
专属性系指在可能存在其他元素杂质、基质和其他来源干扰的情况下,采用的分析方法(包括供试品制备方法和测定方法)能够准确测定供试品中待测元素的能力。根据各测定方法的特性进行专属性试验,通常考察的因素包括试剂空白、供试品基质及其他元素干扰等。当基质干扰影响供试品中待测元素准确定量时,应采取有效措施消除干扰或使干扰降至忽略不计,必要时,选择专属性更好的方法。
(2)线性及范围
应在设计的范围内建立线性关系。制备不少于5个不同浓度水平,相关系数应符合相关要求。范围为分析方法能达到精密度、准确度和线性要求时的高低限浓度或量的区间。
(3)准确度
可在供试品中添加元素杂质,采用回收率试验结果来评价准确度,也可将拟用方法的测定结果与另一良好定义并经验证的方法的测定结果比较,来评价准确度。
采用回收率试验验证准确度时,在供试品中添加一定量的元素标准溶液,根据待测元素限度,设计至少高、中、低3个不同的浓度(通常使添加后的待测元素的量在限度值的50%~150%范围内,当供试品中元素杂质含量较低时,可设计更低的浓度点,如报告阈值水平),每个浓度至少平行制备3份回收率样品,测定并计算回收率。除另有规定外,各元素每个浓度的平均回收率应在70%~150%之间。
(4)精密度
重复性:在供试品中添加一定量的元素标准溶液,使待测元素的量为100%限度值,平行制备6份,或设计不少于3个浓度水平,每个浓度水平至少平行制备3份,测得结果(n≥6或n≥9)的相对标准偏差应不大于20%。
中间精密度:考察随机变动因素对方法精密度的影响。通过不同日期、不同分析人员、不同仪器进行重复性试验,测定结果的相对标准偏差应不大于25%。
重现性:在方法转移或方法复核时,应考察方法的重现性。
(5)定量限与检测限
可选择信噪比法或基于响应值标准偏差和标准曲线斜率法来评估检测限和定量限,湿化学法也可采用直观法来评估检测限。定量限也可通过满足准确度要求来直接验证,定量限应不得大于报告阈值。
(6)耐用性
作为分析方法开发一部分的耐用性试验,在方法验证时,通常不必重复试验,但应予以完善确认,详见分析方法验证指导原则(指导原则9101)。
耐用性通常可考察供试品溶液的稳定性、供试品制备过程中使用的酸、碱等关键试剂的量或浓度、供试品消解程序、仪器分析参数等关键实验因素。
8.4 系统适用性
根据分析方法开发及其验证结果,特别是耐用性考察结果,如有必要且可能,应设立系统适用性试验及其要求,并在品种项下规定。
方法转移和使用时,系统适用性试验应符合规定。
通常,定量测定时,在供试品溶液分析前后均需测定限度浓度的对照品溶液,对照品溶液复测后各元素响应值应为之前对照品溶液响应值的80%~120%;还应测定质控样品,可配制100%限度的回收率样品作为质控样品,测得的回收率应在70%~150%之间。
限度浓度:依据各元素PDE计算供试品中各元素限度,并根据测定需要配制成供试品溶液后形成的浓度。如口服制剂中镉元素PDE为5μg/d,按10g/d的最大日剂量计算,则该制剂中镉元素的限度为0.5μg/g,若取供试品0.5g用50ml溶剂溶解,则该供试品溶液中隔元素的限度浓度为5ng/ml。