0861 残留溶剂
药品中的残留溶剂系指在原料药、辅料或制剂生产过程中使用或产生的,并在实际工艺过程中不能完全除去的有机挥发性化合物。本通则规定了药品中残留溶剂评估与控制的基本原则,以及残留溶剂鉴别、检查与定量测定的方法,用于残留溶剂的测定、评估和控制。
评估与控制
一般原则
选择适当的溶剂可提高产品的收率或决定其晶型、纯度和溶解度等性质,因此,溶剂有时可能是合成工艺的关键因素。由于残留溶剂没有治疗益处,为符合药品的质量标准、生产质量管理规范(GMP)或其他质量要求,应尽可能除去所有残留溶剂。本通则不针对特意用作辅料的溶剂和溶剂化物,但制剂中的这些溶剂也应接受评价,并论证其合理性。
为保护患者的安全,制剂的残留溶剂量不应高于安全性数据可支持的水平。除非在风险-收益评估中能强有力地论证使用这些溶剂的合理性,否则在生产原料药、辅料或制剂时,应避免使用已知会引起不可接受毒性的溶剂(第一类,表1);应限制使用一些毒性较不严重的溶剂(第二类,表2),以防止患者出现潜在的不良反应;如实际可行,应尽可能使用低毒性的溶剂(第三类,表3)。
当已知生产或纯化工艺中出现上述溶剂时,应对残留溶剂进行检查,但仅需要检查原料药、辅料或制剂的生产或纯化中使用的或产生的溶剂。生产企业可选择直接检测制剂中的残留溶剂,也可选择检测制剂生产所用的原料药和辅料等各成分中的残留溶剂。根据制剂生产所用的各成分中的残留溶剂水平,累积计算出制剂中的残留溶剂整体水平;如果计算结果等于或低于本通则规定的接受水平,则无需考虑对制剂进行相关残留溶剂检查;如果计算结果高于规定的接受水平,则应对制剂中相关残留溶剂进行检查,以确定制剂工艺是否将相关溶剂的量降至可接受水平;如果制剂生产中使用了某种溶剂,也应对其在制剂中的残留量进行检查。
本通则规定了药品中可接受的残留溶剂限度。这些限度适用于所有剂型和给药途径。在特定情况下,如短期(30天或更短)用药或局部用药时,可接受更高的残留溶剂水平,应根据不同情况论证这些溶剂水平的合理性。
由于不同生产企业或不同工艺所用溶剂种类可能存在差异,无论各品种正文中是否设置残留溶剂检查项,生产企业均应基于本通则的要求,遵循质量风险管理原则,结合生产工艺,对使用或可能产生的残留溶剂,采用本通则推荐的方法或其他经验证、核准的方法进行测定和控制,使残留溶剂量符合本通则规定的限度要求。
基于风险评估的残留溶剂分类
“每日允许暴露量”(Permitted Daily Exposure,PDE)系指药学上可接受的残留溶剂每日摄入量。根据对人类健康的潜在风险评估,将残留溶剂分为以下三类(表1~表3)。
第一类溶剂:应避免的溶剂
已知的人体致癌物,疑似人体强致癌物,以及环境危害物。
第二类溶剂:应限制的溶剂
非遗传毒性动物致癌物质,或可能导致神经毒性或致畸性等不可逆毒性的溶剂,以及可能有其他严重但可逆的毒性的溶剂。
第三类溶剂:低潜在毒性的溶剂
对人体低潜在毒性的溶剂,无须制定基于健康的暴露限度。第三类溶剂的PDE为每天50mg(50mg/d)或以上。
残留溶剂的限度
1.应避免的溶剂
由于第一类溶剂具有不可接受的毒性或对环境造成危害,原料药、辅料及制剂生产中不应使用该类溶剂。但是,为了生产一种有显著治疗优势的制剂而不得不使用时,除非经过论证,否则应按表1进行控制。1,1,1-三氯乙烷因危害环境而列入表1,其限度1500ppm为基于安全性数据确定。
2.应限制的溶剂
表2所列溶剂由于其固有毒性,应限制其在制剂中的使用。表2中,溶剂的PDE值修约至0.1mg/d,限度值修约至10ppm。
3.低潜在毒性的溶剂
第三类溶剂(表3)可视为低毒、对人类健康危害风险较低的溶剂。第三类溶剂不包括在药品中通常可接受的已知对人类健康有危害的溶剂。虽然许多第三类溶剂缺乏长期毒性或致癌性研究,但现有数据表明,这类溶剂在急性或短期研究中毒性较小,遗传毒性研究结果呈阴性。因此,认为每日摄入50mg(用下述方法1计算时,对应于5000ppm)或更少量时无须论证即可接受。如符合生产能力和GMP的实际情况,也可接受更大的残留量。
4.无足够毒理学数据的溶剂
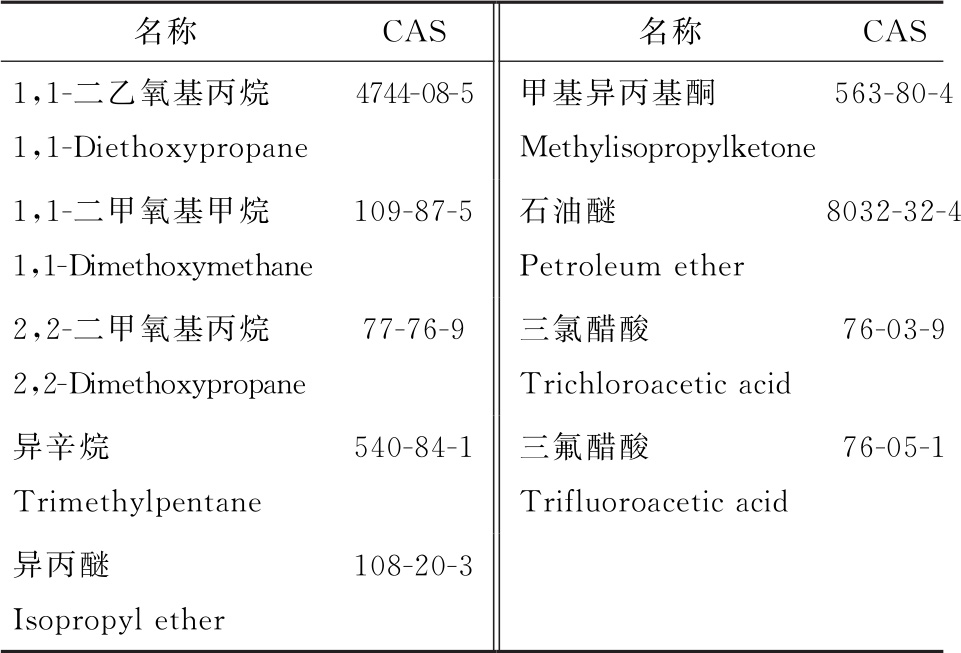
生产企业在辅料、原料药和制剂生产中还可能会使用表4中的溶剂,但尚无足够的毒理学数据,故无PDE值。生产企业应论证这些溶剂在制剂中残留量的合理性。
表中所列的溶剂分类及其建议限度来源于ICH Q3C残留溶剂指导原则,且将随安全性数据更新和ICH Q3C版本更新而变更。
第二类溶剂限度的确定方法
根据PDE值,制定第二类溶剂的限度时有两种方法。
方法1:使用表2中列出的浓度限度(ppm)。这些浓度限度是在假定某制剂的日给药量为10g时,用以下公式(1)计算而得。
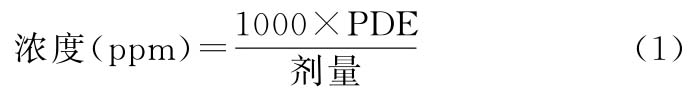
其中,PDE的单位为mg/d,剂量的单位为g/d。
在所有原料药、辅料或制剂中,这些限度被认为是可接受的。因此,若日摄入总量未知或未定,可采用该方法。若处方中的所有辅料及原料药都符合方法1的限度,则这些组分可按任意比例使用。若日摄入总量不超过10g,则无须进一步计算。若制剂的给药剂量超过10g/d,则应考虑采用方法2确定残留溶剂的允许限度。
方法2:认为制剂的各种成分不必都符合方法1的限度。可用表2中注明的PDE(mg/d)、已知最大日摄入总量和公式(1)来确定制剂中允许的残留溶剂的浓度。
如能证明残留溶剂已减少到实际的最低水平,以方法2确定的限度是可以接受的。这些限度在分析精密度、生产能力和生产工艺的合理变更方面应具有可行性,并应反映当前的生产技术水平。
应用方法2时可将制剂各成分(包括原辅料等)所含的残留溶剂累加。每日摄入的溶剂总量应低于给定的PDE值。
残留溶剂的报告方式
制剂生产企业需要了解原料药和辅料残留溶剂的相关信息,以符合本通则的规定。原料药或辅料供应商可参考以下几种示例为制剂生产企业提供相关信息。供应商可视情况选择以下一种。
(1)仅可能存在第三类溶剂。干燥失重小于0.5%。
(2)仅可能存在第二类溶剂,如X、Y……。全部低于方法1的限度(这里供应商可将第二类溶剂用X、Y……来表示)。
(3)可能同时存在第二类溶剂X、Y……和第三类溶剂。残留的第二类溶剂低于方法1的限度,残留的第三类溶剂低于0.5%。
如果可能存在第一类溶剂,应进行鉴别并定量。
“可能存在”系指生产过程的最后一步和生产过程较前几步使用的、用经验证的工艺始终不能除尽的溶剂。
如果第二类溶剂高于方法1的限度或第三类溶剂高于0.5%,应对其进行鉴别和定量。
测定方法
药品中残留溶剂的鉴别、限度检查和定量测定通常采用色谱技术如气相色谱法。可采用本通则推荐的方法,或选择与药品生产特定情况相适应的经验证的分析方法。当仅有第三类溶剂存在时,也可采用经适当验证的非专属性方法进行检查,如干燥失重检查法。验证时应考虑溶剂挥发性对分析方法的影响。
残留溶剂的方法学验证应遵循分析方法验证指导原则(指导原则9101)相关要求。
下述推荐的方法照气相色谱法(通则0521)测定。
色谱柱
1.毛细管柱
一般情况下,极性相近的同类色谱柱可以互换使用。
(1)非极性色谱柱 固定液为100%的二甲基聚硅氧烷的毛细管柱。
(2)极性色谱柱 固定液为聚乙二醇(PEG-20M)的毛细管柱。
(3)中极性色谱柱 固定液为35%二苯基-65%甲基聚硅氧烷、50%二苯基-50%二甲基聚硅氧烷、35%二苯基-65%二甲基聚硅氧烷、14%氰丙基苯基-86%二甲基聚硅氧烷、6%氰丙基苯基-94%二甲基聚硅氧烷的毛细管柱等。
(4)弱极性色谱柱 固定液为5%苯基-95%甲基聚硅氧烷、5%二苯基-95%二甲基聚硅氧烷共聚物的毛细管柱等。
2.填充柱
以直径为0.18~0.25mm的二乙烯苯-乙基乙烯苯型高分子多孔小球或其他适宜的填料作为固定相。
供试品溶液的制备
为使供试品中的残留溶剂得以完全释放,应选择合适的溶剂或通过适当方式使供试品尽可能溶解。如有实验或数据证明其中的残留溶剂已得到最大限度的释放,供试品不能完全溶解也是可以接受的。这种情况可先将供试品粉碎成细粉,但粉碎操作应尽快进行,避免因摩擦生热而使溶剂挥发损失。
精密称(量)取供试品适量,根据供试品和待测溶剂的溶解性能,选择适宜的且不干扰待测溶剂的溶剂,根据残留溶剂的限度要求,制成适当浓度的供试品溶液,以满足定量测定的要求。通常以水为溶剂,对于非水溶性供试品,可采用二甲基甲酰胺、二甲基亚砜或其他适宜的溶剂。
对照品溶液的制备
精密称(量)取待测溶剂适量,采用与制备供试品溶液相同的方法和溶剂制备对照品溶液。用水做溶剂时,如待测溶剂在水中溶解性不好,可先将待测溶剂溶解在50%二甲基亚砜或二甲基甲酰胺溶液中,再用水逐步稀释。
一般根据待测残留溶剂的限度要求确定对照品溶液的浓度。
测定法
第一法(毛细管柱顶空进样等温法)
色谱条件 柱温一般为40~100℃;常以氮气或氦气为载气;顶空瓶平衡温度为70~85℃,顶空瓶平衡时间通常为30~60分钟;进样口温度一般为200℃,如采用火焰离子化检测器(FID),温度一般为250℃。
测定法 取对照品溶液和供试品溶液,分别进样,测定待测峰的峰面积。
第二法(毛细管柱顶空进样程序升温法)
色谱条件 柱温一般先在40℃维持8分钟,再以每分钟8℃的升温速率升至120℃,维持10分钟;以氮气或氦气为载气;顶空瓶平衡温度为70~85℃,顶空瓶平衡时间通常为30~60分钟;进样口温度一般为200℃,如采用火焰离子化检测器(FID),温度一般为250℃。
测定法 取对照品溶液和供试品溶液,分别进样,测定待测峰的峰面积。
第三法(溶液直接进样法)
如采用毛细管柱,可参考以下色谱条件。
色谱条件 柱温可采用等温法或程序升温法,条件参考第一法或第二法;以氮气或氦气为载气;进样口温度一般为200℃,如采用火焰离子化检测器(FID),温度一般为250℃;进样量一般不超过数微升。
测定法 取对照品溶液和供试品溶液,分别进样,测定待测峰的峰面积。
系统适用性试验
根据分析方法验证的结果(特别是耐用性考察结果),可选择设置以下系统适用性试验及其可接受标准。
(1)理论板数:在柱效影响分离效能时,可规定色谱柱应达到的最小理论板数。
(2)分离度:一般情况,待测溶剂色谱峰与相邻色谱峰的分离度应满足分离要求,当分离检测多种残留溶剂且分离度受到挑战时,基于待测峰不受干扰或干扰可忽略不计,可合理规定待测峰与相邻峰的分离度,必要时,可用待测峰与相邻峰的峰谷比来描述分离要求。
(3)对称性:在峰拖尾可能影响待测峰的准确定量或干扰邻近峰测量时,可对色谱峰的对称性提出要求。
(4)灵敏度:可根据残留溶剂的限度浓度或更低的浓度,制备对照品溶液或加标供试品溶液,作为灵敏度试验溶液。通常,定量限的信噪比应不小于10,且不得高于报告阈值;检测限的信噪比应不小于3。
(5)重复性:取对照品溶液或加标供试品溶液,重复进样5~6次,待测物峰面积(或待测物与内标物峰面积之比)的相对标准偏差(RSD)应满足检测的精密度要求。一般情况下,以外标法测定时,待测物峰面积的RSD应不大于10%;以内标法测定时,待测物与内标物峰面积之比的RSD应不得大于5%。
保留时间和相对保留时间常用于评价系统适用性,如在品种项下列出但未明确为系统适用性要求,它们仅作为一种参考。
在整个分析过程中,色谱系统应满足规定的系统适用性要求,确保实验结果可被接受。
以上推荐的色谱条件包括顶空条件等实验参数,可能因设备和待测溶剂而异,在建立适合于特定供试品中的残留溶剂测定法时,可对推荐的实验参数进一步优化,优化后的实验参数经验证后,在品种的相关控制项目中进行描述。在方法实际应用中,为满足系统适用性要求,可按气相色谱法(通则0521)色谱参数调整的有关规定对品种项下的色谱条件作适当调整。
残留溶剂的鉴别
对于未知残留溶剂,常用的鉴别方法主要有但不限于:
(1)保留时间
在相同的色谱条件下,待测溶剂峰的保留时间与对照品溶液相应色谱峰的保留时间一致,可用于鉴别待测物中的残留溶剂。两个保留时间不同的色谱峰归属于不同化合物,但两个保留时间一致的色谱峰有时未必可归属为同一化合物,在作未知物定性分析时应特别注意。
采用测定法中第一法或第二法的色谱条件,或其他经验证的方法,在确保检测灵敏度、分离度等参数满足相关技术要求的情况下,选择两种不同极性的色谱柱(如非极性、中极性或极性色谱柱)试验。当待测溶剂峰在不同极性色谱系统中的保留时间均与对照品溶液色谱峰保留时间一致时,可初步鉴别供试品中可能存在的残留溶剂。
(2)利用质谱检测器提供的质谱信息
气相色谱质谱联用仪的质谱检测器能提供与色谱峰对应的残留溶剂分子质量和结构特征等更多、更可靠的信息,不仅可用于已知残留溶剂的鉴别,还可提供未知残留溶剂的结构信息(通则0431)。
利用质谱信息库检索进行未知化合物结构和定性分析时,通常是将测得的谱图信息简化为少数最有意义的峰,然后再和质谱信息库中的对应谱图相比较。通过质谱信息库检索可对未知残留溶剂结构进行确证,或为需要结构确证的残留溶剂提供充分的证据。
残留溶剂的检查和定量
(1)限度检查 以内标法测定时,供试品溶液所得待测溶剂峰面积与内标峰面积之比不得大于对照品溶液的相应比值。以外标法测定时,供试品溶液所得待测溶剂峰面积不得大于对照品溶液的相应峰面积。
(2)定量测定 按内标法或外标法计算各残留溶剂的量。
对于第三类溶剂的测定。如果已知供试品中仅存在第三类溶剂,可采用干燥失重测定法(通则0831)进行测定。但当干燥失重大于0.5%,或供试品中存在其他溶剂时,应采用上述推荐方法或其他经验证的方法对供试品中的第三类溶剂进行鉴别,并根据需要进行限度检查或定量测定。
残留溶剂的分析策略图见图1。
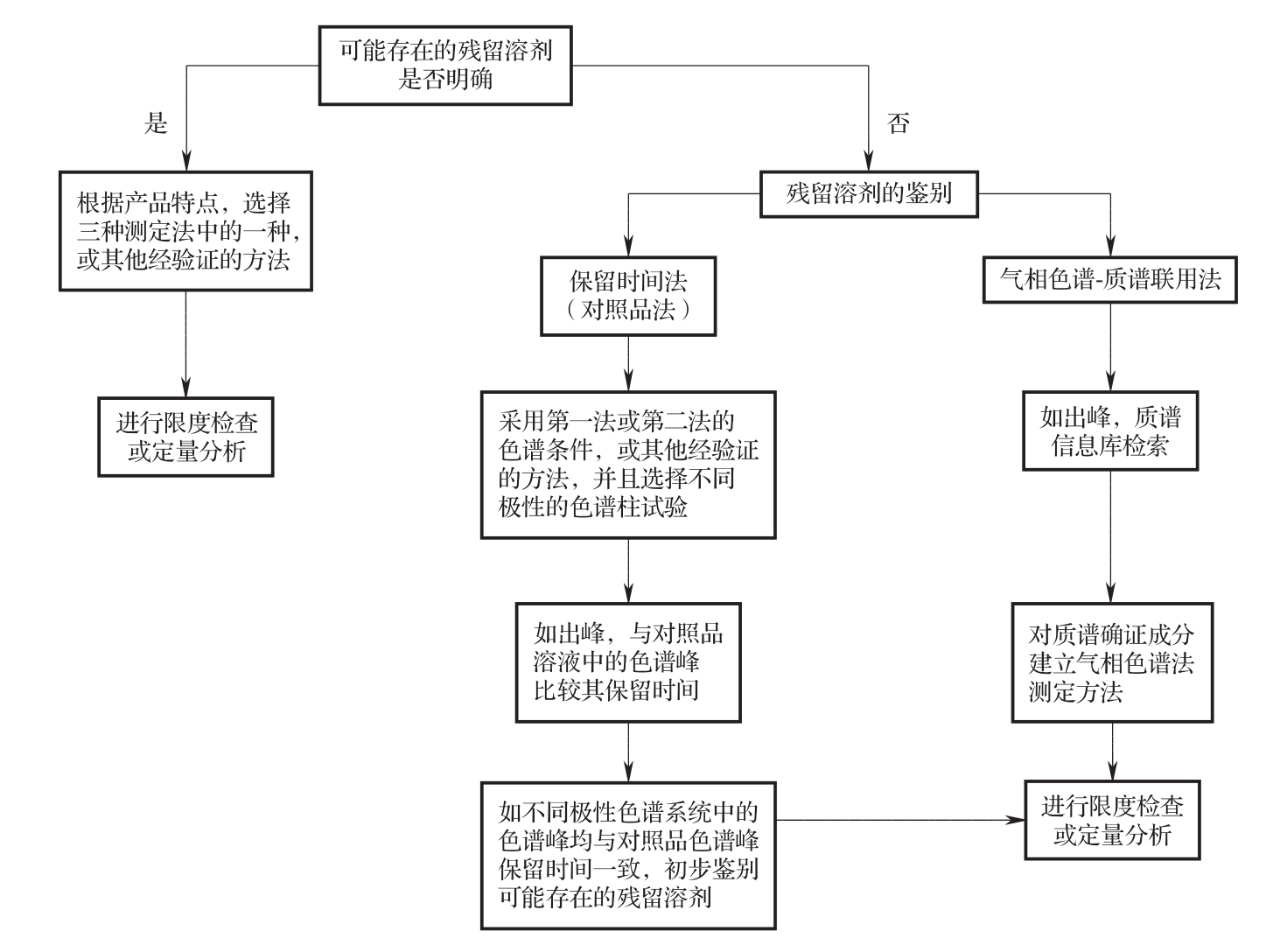
图1 分析策略图
分析方法建立和使用中的其他考虑
(1)当需要检查的残留溶剂种类不多,且极性差异较小时,可采用等温法。当需要检查的残留溶剂种类较多,且极性差异较大时,可采用程序升温法。
(2)顶空条件的选择
①应根据供试品中残留溶剂的沸点选择顶空平衡温度。对于沸点较高的残留溶剂,通常选择较高的平衡温度;但此时应兼顾供试品的热分解特性,尽量避免供试品产生的挥发性热分解产物对测定的干扰。
顶空平衡温度一般应低于溶解供试品所用溶剂的沸点10℃以下,能满足检测灵敏度即可。过高的平衡温度,可使顶空瓶的气密性变差,导致定量准确性的降低。
②顶空平衡时间一般为30~60分钟,以保证供试品溶液的气-液两相有足够的时间达到平衡。不同的测定体系所需的平衡时间不同,需根据实际考察确定合适的平衡时间,既保证供试品溶液达到气-液平衡,又不会因为顶空时间过长导致顶空瓶气密性变差而降低定量准确性。
③对照品溶液与供试品溶液必须使用相同的顶空条件。
(3)对于沸点较高的甲酰胺、2-甲氧基乙醇、2-乙氧基乙醇、乙二醇、N-甲基吡咯烷酮、环丁砜、二甲基甲酰胺、二甲基乙酰胺等第二类溶剂,不易通过顶空进样测定获得满足要求的灵敏度,可使用其他经验证的方法进行测定。
(4)含氮的碱性残留溶剂测定 普通气相色谱仪中的不锈钢管路、进样器衬管等对有机胺等含氮碱性化合物具有较强的吸附作用,导致其检出灵敏度降低,应采用惰性的硅钢材料或镍钢材料管路;采用溶液直接进样法测定时,供试品溶液应不呈酸性,以免待测溶剂与酸反应后不易汽化。
通常采用弱极性的色谱柱或其他填料预先经碱处理过的色谱柱分析含氮碱性化合物,如采用胺分析专用柱进行分离,效果更好。
对不宜采用气相色谱法测定的含氮碱性化合物,可采用其他方法如离子色谱法等测定。
(5)含羧酸的酸性残留溶剂测定 含羧酸的酸性残留溶剂如甲酸、醋酸等,采用气相色谱法测定一般响应值低,可通过酯化衍生等样品处理方式提高检测灵敏度,也可采用液相色谱法或其他适宜的方法测定。采用液相色谱法测定时,流动相和供试品溶液应呈酸性。
(6)流速的选择 流速一般与所用的色谱柱内径相适应,可根据系统适用性试验情况选择合适的流速。
(7)检测器的选择 对含卤素的残留溶剂如三氯甲烷等,采用电子捕获检测器(ECD),易获得较高的灵敏度。
(8)干扰峰的排除 供试品中未知杂质或其挥发性热降解物易对残留溶剂的测定产生干扰。干扰作用包括在测定的色谱系统中未知杂质或其挥发性热降解物与待测物的保留时间相同(共出峰);或热降解产物与待测物的结构相同(如甲氧基热裂解产生甲醇)。当测定的残留溶剂超出限度,但未能确定供试品中是否有未知杂质或其挥发性热降解物对测定有干扰作用时,应通过试验排除干扰作用的存在。对第一种干扰作用,通常采用2种极性不同的色谱系统对相同的供试品进行测定,比较不同色谱系统的测定结果。如两者结果一致,则可以排除测定中有共出峰的干扰;如果两者结果不一致,则表明测定中有共出峰的干扰。对第二种干扰作用,通常通过测定已知不含该溶剂的对照样品来加以判断。
(9)定量方法的验证 当采用顶空进样时,供试品与对照品处于不完全相同的基质中,故应考虑气液平衡过程中的基质效应(供试品溶液与对照品溶液组成差异对顶空气液平衡的影响)。由于标准加入法可以消除供试品溶液基质与对照品溶液基质不同所引起的基质效应的影响,故通常采用标准加入法验证定量方法的准确性;当标准加入法与其他定量方法的结果不一致时,以标准加入法的结果为准。
(10)不同实验室在测定同一供试品时,可能会采用不同的试验方法,当测定结果处于合格与不合格的边缘时,以内标法或标准加入法的结果为准。
表1 第一类溶剂
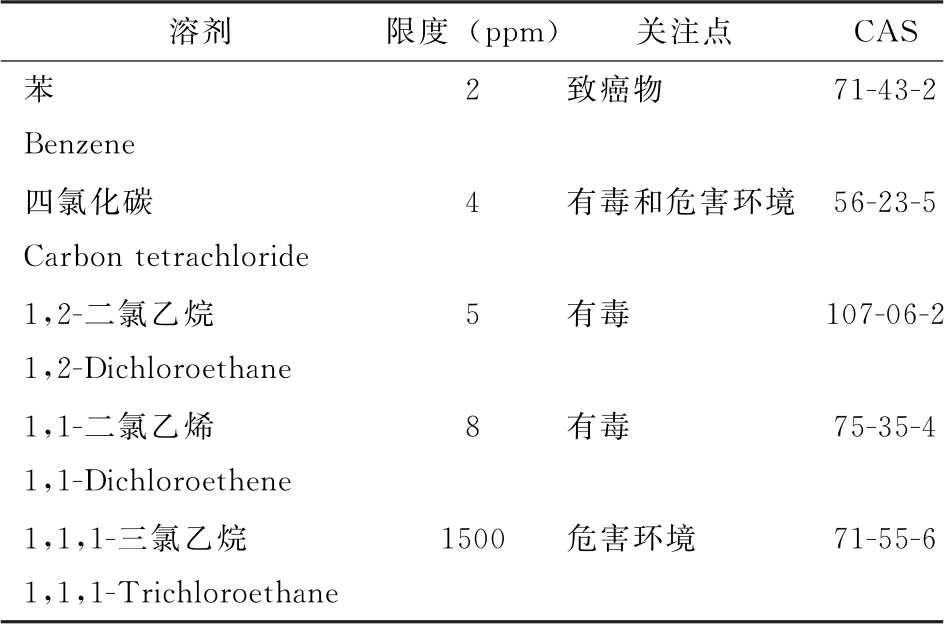
表2 第二类溶剂
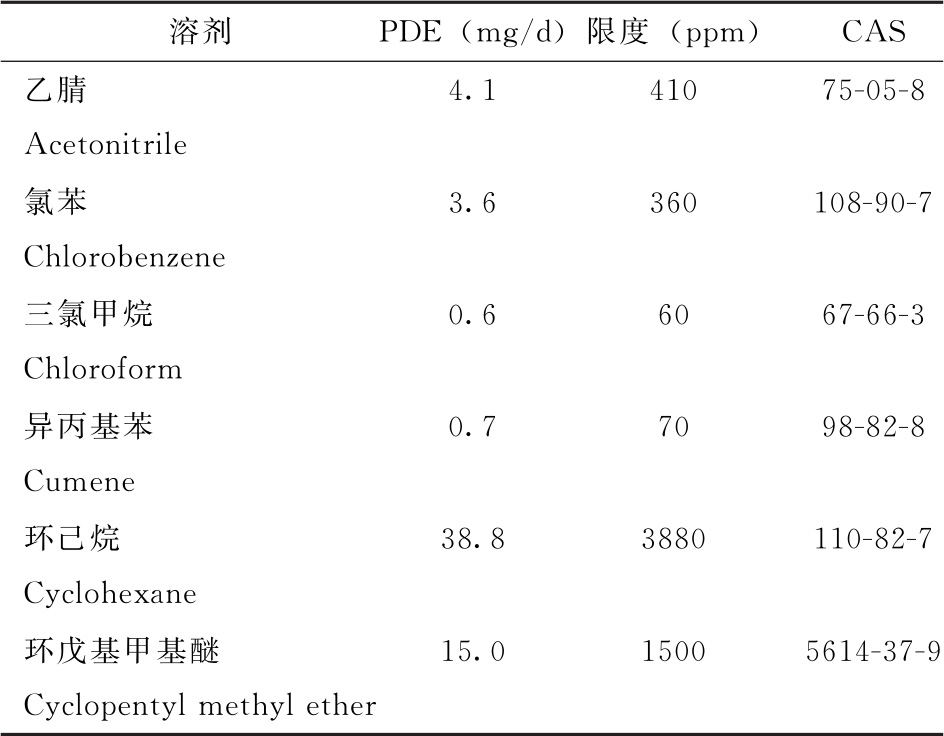
续表
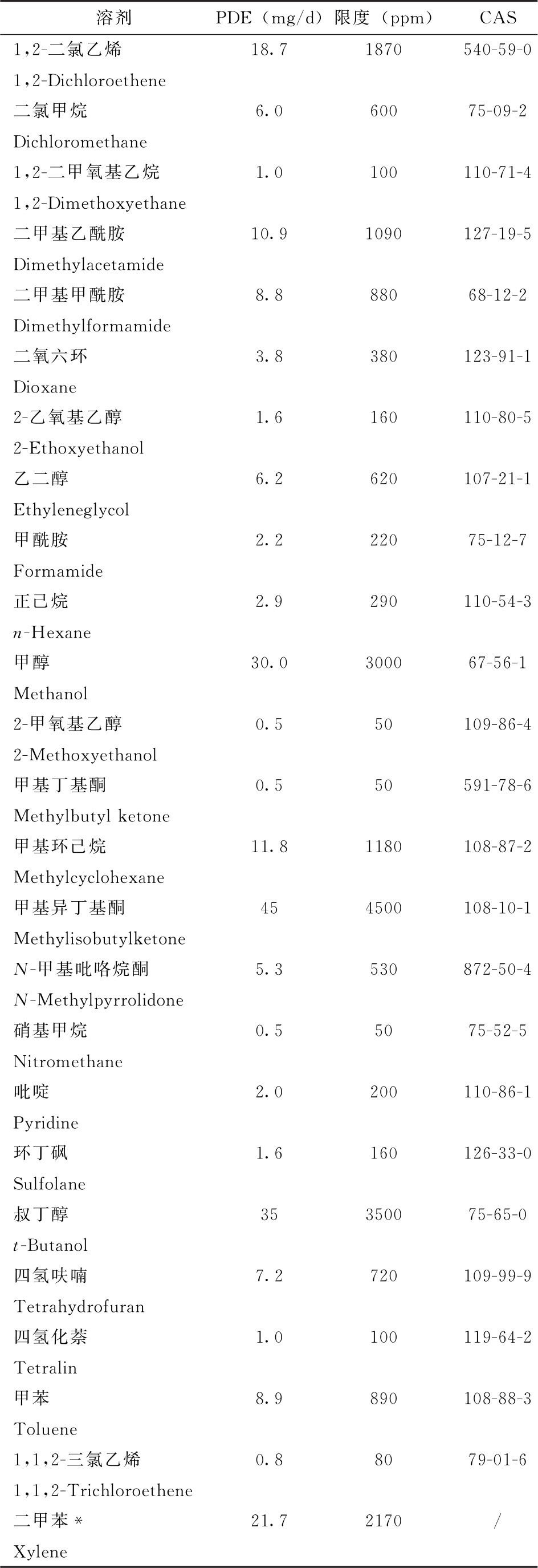
注:*通常为60%间二甲苯,14%对二甲苯,9%邻二甲苯和17%乙苯
表3 第三类溶剂

表4 无足够毒理学数据的溶剂