
新药制剂中的杂质
Step 5
2006/6/2
新药制剂中的杂质
ICH三方协调指导原则
2003年2月5日在ICH指导委员会会议上进入ICH进程第四阶段,并推荐给ICH三方管理部门采纳。
2006年6月6日对附件2作了修订。
目录
介绍
1.1 本指导原则的目的
1.2 背景
1.3 本指导原则的适用范围
降解产物报告和控制的合理性
分析方法
各批次产品降解产物报告的内容要求
质量标准中的降解产物检查项目
降解产物的界定
术语
附件1:新制剂中的降解产物限度
附件2:在申报资料中如何根据鉴定或界定限度的要求报告杂质的检测结果示例
附件3:降解产物鉴定和界定的决策树
新药制剂中的杂质
1.介绍
1.1本指导原则的目的
本文件为新药制剂在申报资料中,对杂质研究的内容和限度确定提供指导,制备该新药制剂所用的化学合成的新原料药尚未在这些地区或成员国注册过。
1.2背景
本指导原则为ICH Q3A(R)“新原料药中的杂质”指导原则的附件,“新原料药中的杂质”应被视作基本准则;如必要,也应遵循ICH Q3C“残留溶剂”指导原则。
1.3本指导原则的适用范围
本原则仅阐述新药制剂中原料药的降解产物或原料药与赋形剂和/或包装容器的反应产物(本原则统一称作“降解产物”)。一般情况下,存在于新原料药中的杂质在新药制剂中不需要监控,除非它们也属于降解产物(见ICH Q6A“质量标准”指导原则)。
本原则不包括从新药制剂的赋形剂或从包装容器渗出产生的杂质,也不适用于临床研究开发阶段的新药制剂。以下制剂不在本原则指导范围内:生物及生物技术产品、缩肽、寡聚核苷酸、放射性药物、发酵制品及其半合成品、草药和来源于动植物的粗制品。另外也不包括:⑴外源性污染物,不应该存在于新药制剂中,可以用GMP来控制的;⑵多晶型;⑶对映体杂质。
2. 降解产物报告和控制的合理性
申报者应对新药制剂的生产和/或稳定性考察中所发现的降解产物进行综述。该综述应包括对制剂中可能的降解途径和因与赋形剂和(或)包装容器反应所产生杂质的科学评价。另外,申报者还要对新药制剂降解产物检测的所有实验室研究工作进行总结,也包括研发过程中生产的批次和采用拟上市工艺生产批次的试验结果。应对不属于降解产物的杂质(如来自于原料药的工艺杂质和由赋形剂产生的杂质)进行说明。应将采用拟上市工艺生产的有代表性批次中的杂质概况与用于研究开发的批次作比较,它们之间存在的任何差异都应进行讨论。
在推荐的放置条件下进行的稳定性考察中发现的任何降解产物,如果大于(>)附件1中给出的鉴定限度限度时,则应对其作结构确证。若无法确证某一降解产物结构时,也应在申报资料中说明实验室对鉴定该物质已作过的所有研究工作。
对不大于(≤)限度限度的降解产物通常不需要确证其结构。但是,对于那些可能有不寻常功效或产生毒性药理作用的降解产物,即使不大于(≤)鉴定限度限度,仍应建立分析方法。在特殊情况下,可以根据在拟上市工艺生产中所获得的经验,及一些技术因素(如:最大产量,原料药相对于赋形剂的低比例,或使用源自于动植物粗制品的赋形剂)对限度限度作合理的调整。
3.分析方法
申报资料文件中应有证据证明分析方法是经过验验证并适用于降解产物的定性和定量检测(见ICH Q2A和ICH Q2B分析验验证指导原则)。尤为重要的是,应能证明分析方法具有检测特定或非特定降解产物的专属性。必要时,还应包括对放置在相对强烈条件(光、热、湿、酸/碱和氧化)下的样品所进行的分析方法验证。当分析方法揭示除降解产物以外还存在其他色谱峰(如:原料药,原料药合成时引入的杂质,赋形剂和由赋形剂产生的杂质),这些峰需在色谱图中进行标注,在验证文件中应探讨他们的来源。
分析方法的定量限应不大于(≤)报告限度限度。
降解产物的量可以用一系列的技术手段测定,包括诸如降解产物本身的响应值与合适的参比对照品的响应值之间的比较,或与新原料药自身的响应值相比较。在控制降解产物的分析方法中所用到的参比对照品应按它们所用的目的进行评估和鉴定。原料药可用于估测降解产物的量。当响应因子不接近时,如果应用校正因子或实际高估降解产物的话,则仍可用该方法。用于已鉴定或未鉴定的降解产物的认可标准和分析方法常常是基于分析方法学上的假设(如:检测响应相同),这些假设是否合理应在申报资料中进行讨论。
对研制开发期间和拟上市生产的制剂所采用的分析方法如有差异,亦应进行讨论。
4.各批次产品降解产物报告的内容要求
申报资料中应提交用于临床、安全性、稳定性试验的所有相关批次以及采用拟上市工艺生产的有代表性批次的新药制剂的分析结果。定量测定结果应数字化,不应使用类似“符合规定”、“符合限度”等一般性术语。应报告新药制剂相关批次中检测到的任何大于(>)报告限度限度(见附件1)的降解产物以及总的降解产物,并附所用分析方法。小于1.0%时,结果报告至小数点后两位(如:0.06%);大于和等于1.0%时,应报告至小数点后一位(如:1.3%)。结果应按常规方法修约(见附件2),建议用数据表格(如:电子表格)。降解产物应用编号或适当的描述表示(如:保留时间)。如要采用较高报告限度限度,则需经充分论证。所有大于(>)报告限度限度的降解产物都应进行累加并以杂质总量报告。
申报资料中应提供代表性样品批次的有标记峰的色谱图(或采用其他方法获得的相关数据),包括在分析方法验证中和长期、加速稳定性研究中所得到的色谱图。申报者应能确保,如管理部门需要,可提供每个批次的完整的降解产物概况(如:色谱图)。
申报资料中应提供每一批次新药制剂的详细信息,具体内容如下:
批号、规格和批量
生产日期
生产地点
生产工艺
直接接触的包装容器
降解产物的含量,单个的和总量
批次的用途(如:临床研究、稳定性研究)
分析方法所用的对照品
用于该制剂的原料的批号
稳定性研究的放置条件
5.质量标准中的降解产物检查项目
新药制剂的质量标准中应包括在上市产品生产和推荐的贮藏条件下预期会出现的降解产物的检查项目。稳定性研究、降解途径的了解、产品开发研究以及实验室研究都可用来确定降解的概况。新药制剂质量标准中列入的降解产物检查项应根据拟上市工艺生产的批次中发现的降解产物来确定。本原则中所指的“特定降解产物”是指列入新药制剂质量标准中有特定限度要求的各个降解产物。特定降解产物可以是结构已确证和未确证的。应对在安全性、临床研究批次和稳定性研究中观察到的降解产物状况、并结合拟上市工艺生产的产品批次中降解产物的状况,综合进行讨论后,再对质量标准中列入和不列入哪些降解产物的理由进行说明。质量标准中应包含特定的已鉴定的降解产物和估计含量大于(>)附件1中给出鉴定限度限度的特定的结构未鉴定的降解产物。对于被认为具有特殊功能或产生药理毒性或未预料到的药理作用的降解产物,其分析方法的定量限/检测限必须与降解产物被控制的量相当。对于未鉴定的降解产物,应明确阐述建立降解产物水平所使用的方法和定量的假设。特定的未鉴定的降解产物应用适当的方法来标示(如:“未鉴定杂质A”,“相对保留时间为0.9的杂质”)。对于任何一个非特定的降解产物应有一个不大于(≤)鉴定限度限度(附件1)的认可标准,对总降解产物也应建立一个认可标准。
对于指定的降解产物,制订其认可标准时,应考虑其在原料药中的认可标准(如有的话),它通常的含量以及它在稳定性研究中、建议的有效期和推荐的贮存条件下的增加量。当然,认可标准的设定不得高于该降解产物经界定的安全含量。
如果没有安全性问题,降解产物的认可标准应建立在来自拟上市工艺生产的新药制剂各批次获取的数据基础上,并允许留有足够的余地以适应常规的生产、分析方法和稳定性方面的正常变异。虽然常规生产的变化是可预测的,但批与批之间降解产物水平的显著变化则可能意味着新药制剂的生产过程尚未能得到很好的控制和验证(见ICH Q6A原则质量标准:决策树#2,新药制剂中特定降解产物认可标准的制定)。
在本指导原则中,限度限度的两位小数点应用(见附件1)并不必然代表特定的降解产物和总的降解产物认可标准的精度。
总之,新药制剂质量标准中应包括以下降解产物的检查项:
每种特定的、已鉴定的降解产物
每种特定的未鉴定的降解产物
任何不大于(≤)鉴定限度限度认可标准的非特定降解产物
降解产物总量
6.降解产物的界定
杂质的界定是获得和评价某些数据的过程,这些数据可用于确保单个杂质或在特定的含量下的一系列杂质的生物安全性。限度限度申报者对所建立的降解产物认可标准应提供包括安全性研究在内的依据。对于一个通过充分的安全性研究和临床研究的新药制剂,其中任何一个降解产物的水平即被认为是已经通过了界定,是合理的。因此关于安全性和/或临床研究期间所用的相关批次产品中降解产物的实际含量的任何资料都是有用的。对于是动物和/或人体中的重要代谢物的那些降解产物,也认为已通过界定。如果安全性实验中实际所给的剂量大于新药制剂临床设计剂量,即使降解产物的量较高也是安全的。在论证这些高含量的合理性时,应考虑如下因素:(1)该降解产物的量在先前的安全性和/或临床研究中也存在并被认为是安全的;(2)降解产物增长量;(3)其他安全性因素。
如果建立的认可标准超过附件1中所给出的界定限度限度,而所获得的试验数据不能用来证明降解产物的认可标准限度是合理的,则必须进行进一步研究(见附件3 )。
根据科学原理和药品的类别及临床使用情况,某些新药制剂降解产物的界定限度可以订得更高或更低一些。例如,当有证据表明这些降解产物先前和病人的副反应有关联的时候,其界定尤其重要。在这种情况下,相对低的界定限度可能是合适的。反之,如果考虑相似情况(病例数、药物类别、临床情况等)后,对安全性的考虑比通常情况小,那么这些药物的界定限度限度可以高一些。对限度的调整应具体情况具体对待。
“降解产物鉴定和界定决策树”(见附件3 )描述了当降解产物的水平超过界定限度限度时应考虑的事项。在某些情况下降低降解产物的含量使其低于限度要比提供降解产物的安全性数据来得简单。或者当文献资料中对某一降解产物的安全性数据比较充分时,也可用于界定该降解产物的限度。如果两者均不可行,则应考虑进行额外的安全性试验。限度如何合理地界定一个降解产物的研究将取决于许多因素,包括病例数、每日剂量、给药途径与疗程。虽然有时候采用分离出来的降解产物进行安全性研究更为合适,但通常是用含有需被控制的降解产物的新药制剂或物质来进行研究。
虽然本指导原则在临床研究开发阶段并不适用,但在开发阶段后期,本原则中的限度限度也适用于评价用拟上市工艺生产的各批次新药制剂中发现的新的降解产物。在开发阶段后期所发现的任何新的降解产物,如果其含量大于(>)附件1中给出的鉴定限度限度,都应鉴定其结构(见附件3 中“降解产物鉴定和界定决策树”)。同样,如果其含量大于(>)附件1中给出的界定限度限度,都应界定其安全性。
安全性研究应将含有代表性水平的降解产物的新药制剂或原料药的安全性试验结果与以前已界定过的物质作比较,也可用被分离出来的降解产物进行研究。
7.术语
降解产物(Degradation Product):是指新药制剂在生产和贮藏过程中,因如光照、温度、pH、水或与赋形剂和/或包装系统互相反应而导致原料药发生化学变化而产生的杂质。
降解概况(Degradation Profile):对新原料药或制剂中被检测到的降解产物的的数量及含量。
开发研究(Development Studies):对制剂生产工艺的放大、优化、验证研究。
鉴定限度(Identification Threshold):为一限度,高于此限度的降解产物需鉴定其结构。
已鉴定的降解产物(Identified Degradation Product):化学结构已明确的降解产物。
杂质(Impurity):新药制剂中除了原料药或赋形剂以外的任何其他成分。
杂质概况(Impurity Profile):对存在于药物制剂中的所有已鉴定和未鉴定的杂质的数量与含量。
新原料药(New Drug Substance):先前尚无在某一地区或成员国注册的具有治疗作用的活性部分(也称为新分子或新化学实体)。它可以是某种已获批准的药物的一种复合物、简单的酯或盐。
界定(Qualification):是获得和评价某些数据的过程,这些数据可用于确保单个杂质或在特定的含量下的一系列杂质的生物安全性。
界定限度(Qualification Threshold):为一限度,高于此限度的降解产物需界定其安全性。
报告限度(Reporting Threshold):为一限度,高于此限度的降解产物需报告其含量。
特定的降解产物(Specified Degradation Product):已被列入新药制剂质量标准中并规定了认可标准的降解产物。一个特定的降解产物可以是结构已经鉴定或未鉴定的。
未鉴定的降解产物(Unidentified Degradation Product):未确证其结构,仅通过定性分析手段(如色谱保留时间等)来定义的降解产物。
非特定的降解产物(Unspecified Degradation Product):新药制剂质量标准中,其限度在总的认可标准中控制而不单独控制的降解产物。
附件1:新药制剂中降解产物的限度
报告限度

鉴定限度

界定限度

附件1注:
1.每日服用原料药的量。
2.降解产物的限度既可用原料药的百分比表示,亦可用每日摄入降解产物的总量表示。若降解产物具异常毒性,则限度应较低。
3.较高限度应进行科学论证。
新药制剂中降解物质在每日最大剂量中所占分量的鉴定、界定和报告阈值图解

标尺放大后的图解
注1:实际限度参照本附件前表
附件2:申请中对降解产物进行鉴定和界定报告的实例
每日最大剂量50mg
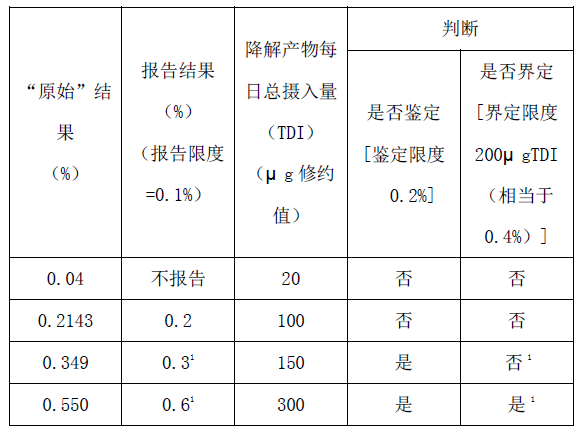
每日最大剂量1.9g
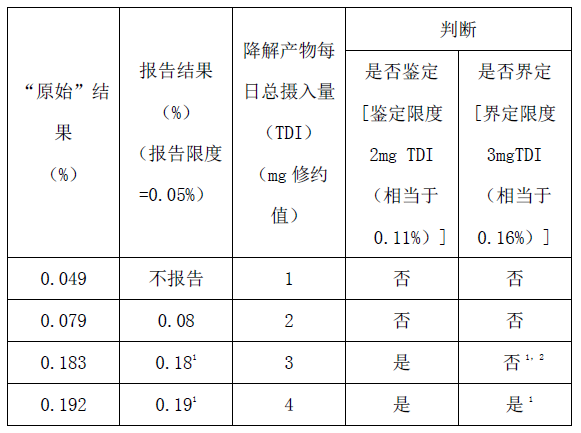
附件2注:
1)鉴定后,如果确定的响应因子和原假设明显不同,应重新检测该降解产物的实际含量,并根据界定限度(见附件1)重新考虑下一步的研究工作。
2)为确证杂质含量是否超过限度,报告的结果应该根据以下原则进行评估:当限度用%表示时,报告的结果应修约到与限度的小数点位数相同后直接与该限度相比较。当限度用TDI表述时,报告值应转换为TDI,并修约至与限度小数点位数相同,并与该限度相比较。例如,降解产物量0.18%相当于3.4 mg(绝对值)TDI,它被修约到3 mg,因此用TDI(3 mg)表示的界定限度是不超标的。
附件3:降解产物鉴定和界定决策树
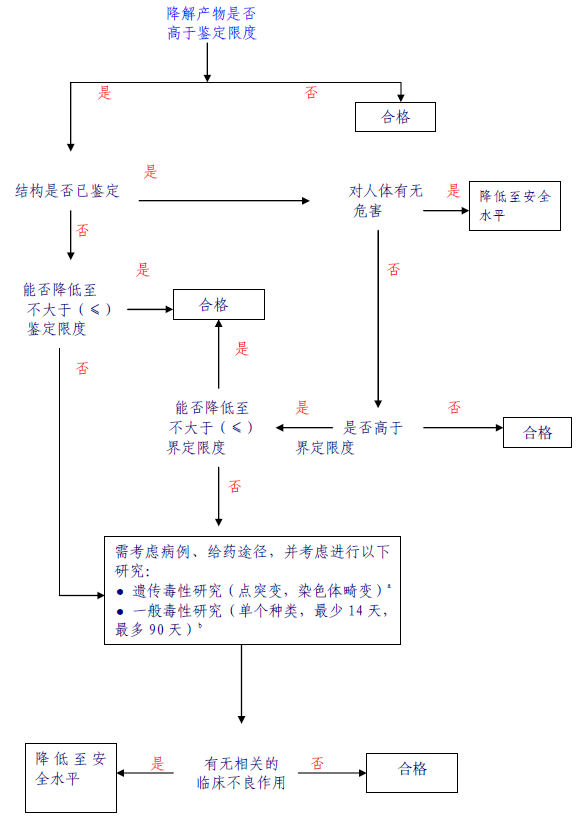
附件3注:
a)如需要,应进行最低限度(如潜在遗传毒性)筛选试验,进行体外点突变和染色体畸变试验被认为是恰当的最低限度筛选试验。
b)如需进行一般毒理研究,应设计一个或多个研究方案,以将未界定的物质与界定的物质进行比较。研究时间应根据可用的相关信息而定,并使用最能反映某一降解产物毒性的动物种属,根据个案分析的原则,可进行单剂量给药试验,尤其是对单剂量给药的药物。一般最短14天,最长90天。
c)如果降解产物具异常毒性,采用较低的限度。
d)例如:已知的该降解产物的安全性数据或其结构的分类是否排除了人接触该浓度杂质的可能?
(c)蒲标网 - 中国药典、药品标准、法规在线查询 ( 津ICP备15007510号 )